


Abstract
Background/Aim: Adenoid cystic carcinoma (SGC) is a common type of salivary gland cancer (SGC). Surgery is the first treatment choice because chemoradiotherapy is usually not effective. Therefore, new treatment modalities are urgently needed. In this study, it was investigated whether the estrogen axis could be a treatment target or not. Materials and Methods: Adenoid cystic carcinoma (ACC) ACCM cells, were used. The specific cell line lacks estrogen receptor (ER). ER was introduced in ACCM cells, and the effect of 17β-estradiol (E2) was investigated on cell proliferation, cell-cycle distribution, and cell motility. Results: E2 induced cell proliferation, and the S-phase fraction increased in a dose-dependent manner. Cell motility was also up-regulated compared to control cells. Conclusion: The estrogen/ER system up-regulated malignant phenotypes in ER-positive ACC, and hormone therapy may have a potential as effective treatment for this malignancy.
Aggressive cancers develop in a multistep process involving genetic and epigenetic alterations. Identifying these changes is essential for understanding the mechanisms of cancer progression, and will enable development of more effective methods of diagnosis and treatment. Human salivary gland cancer (SGC) is a relatively slow-growing neoplasm of secretory glands that occurs most frequently in the minor and major salivary glands (1, 2). However, SGC also includes highly aggressive tumors that invade adjacent tissues and metastasize to distant organs at an early stage (1, 3). Adenoid cystic carcinoma (ACC) is among the most common malignant SGC types, with a very low 10- to 20-year survival rate (3-6). Recurrent cases of ACC are particularly difficult to manage due to ineffectiveness of radio- and chemotherapy and the difficulty of performing wide surgical resection for cosmetic reasons and due to anatomical limitations (7, 8). Therefore, new treatment approaches for SGC are urgently needed.
Sex steroid hormones such as estrogen and progesterone (Pg) play an important role in normal mammary gland development and along with their receptors, influence breast cancer progression (9). Morphological mimicry of breast tumors by SGC with respect to histology and steroid hormone receptor status is a well-known phenomenon in the field of surgical pathology (10). Moreover, SGC occurs more frequently in females (1, 2), and salivary gland and breast tissue ACC show remarkable similarities (10). Certain studies have suggested the possibility of steroid hormone receptor involvement in SGC progression (2, 3, 7).
The Pg receptor (PgR) system has been implicated in the development of malignant phenotypes in breast cancer (11, 12). And we have mentioned that this system was strongly associated with malignant phenotypes of ACC (13). The estrogen-estrogen receptor (E2-ER) system is critically involved in breast cancer (14-18) and may modulate SGC progression along with the Pg-PgR system. This was investigated in the present study in order to assess the therapeutic potential of hormones for the treatment of SGC.
Materials and Methods
Cell culture. The human ACCM cell line, derived from salivary gland ACC (19), was purchased from the Cell Bank of the Type Culture Collection of the Chinese Academy of Science (Shanghai, China). T47D and MDA231 human breast cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Roswell Park Memorial Institute 1640 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 5% charcoal-stripped fetal bovine serum (FBS) at 37°C in an atmosphere of 5% CO2. ACCM cells lack ER and PgR; the estrogenic effects of phenol red were therefore disregarded in this study.
Introduction of ER in ACCM cells. The pSG5-ER plasmid encoding human ER was a gift from Prof. P. Chambon (Institute of Genetics and Molecular and Cellular Biology, Strasbourg, France). The pBK-CMV vector (Stratagene, La Jolla, CA, USA) containing the neomycin resistance gene was cotransfected with pSG5-ER into ACCM cells using Lipofectamine Plus reagent (Life Technologies, Carlsbad, CA, USA). Neomycin-resistant clones were selected with medium containing G418. Cells co-transfected with empty pSG5 and pBK-CMV vectors served as a control. ACCM cells expressing ER were pooled and after screening ER expression level, three ACC-ER clones with various expression levels of ER were produced.
Chemical treatment. E2 (17β-estradiol) was obtained from Sigma and prepared as a 10-mM stock solution in ethanol (Et). For most experiments, E2 was used at a concentration of 10 nM in 10 ml culture medium for a final ethanol concentration of 0.1%. Control cells were treated with E2 or 0.1% ethanol only. Cells were treated with ethanol or E2 once daily.
Western blot analysis. Cells were lysed in 2× Laemmli buffer and stored at −70°C. Protein concentration was determined using the DC Protein Assay kit (Bio-Rad, Hercules, CA, USA). Samples (20-30 μg of total protein) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Hybond P; Amersham Biosciences, Little Chalfont, UK). Membranes were blocked for 1 h at room temperature with Tris-buffered saline with Tween-20 (20 mM Tris, 137 mM NaCl, 3.8 mM HCl, and 0.1% Tween-20) containing 5% nonfat milk, and blots were probed with anti-ERα (EP1; Dako, Glostrup, Denmark), anti-ID1 (Z-8; Santa Cruz Biotechnology, Santa Cruz, CA, USA), or anti-actin (C4; Chemicon International, Temecula, CA) antibodies for 1 h. Membranes were washed and incubated with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA), washed, and developed by enhanced chemiluminescence using the Amersham ECL-Plus kit according to the manufacturer's instructions.
Flow cytometry. The distribution of the cell cycle was measured by flow cytometric analysis. Samples for flowcytometric analysis were prepared from the cultured cells using a Cycle TESTTM PLUS DNA reagent kit (Becton Dickinson Immunocytometry Systems, Ventura, CA, USA). Flowcytometric analysis was performed in a FACScalibur flowcytometer equipped with a Cell QUEST software program (Becton Dickincon & Co., Mountain View, CA, USA). Analysis was carried out on 2×104 nuclei per sample.
Cell proliferation assay. The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) method was used to measure the proliferation of SGCs. The control and ID1AS-transfected SGCs were placed into 96-well plates (1×104 cells/ml) for 48 h, and 20 μl of MTS solution were added to each well for 4 h at 37°C. Absorbance was measured at 490 nm using iMark micro plate reader (Bio-Rad).
PCR-based telomerase assay and reverse transcriptase (RT-)PCR. The telomeric repeat amplification protocol assay was performed as previously described (20), with minor modifications. Protein was extracted from cells cultured in medium containing 5% serum for 24 h in the presence of ethanol or E2; 0.6 mg protein extract was analyzed. An internal control was used for semiquantitative estimation of telomerase activity levels and identification of false-negative tumor samples containing Taq polymerase inhibitors. A 15-μl aliquot of PCR product was resolved by electrophoresis on a 12% non-denaturated polyacrylamide mini gel, which was stained with SYBR Green 1 nucleic acid gel stain (diluted to 1:10,000; FMC Bioproducts, Rockland, ME, USA) for 30 min. The gel was visualized under ultraviolet transillumination.
Total RNA (3 μg) was obtained from cells cultured in medium containing 5% serum for 24 h in the presence of ethanol or E2 (10 nM). Primer sequences for amplifying human telomerase reverse transcriptase (hTERT), the catalytic subunit of telomerase, as well as PCR conditions have been previously described (21). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as an internal control for the reaction. A 10-μl aliquot of the PCR products was resolved by electrophoresis on a 1% agarose gel stained with ethidium bromide. The expression of hTERT mRNA was determined relative to that of GAPDH.
Colony formation assay. Assays were performed as described previously (22). Briefly, liquefied 2% agarose was mixed with an equal volume of 2X DMEM/F12 growth medium lacking serum and supplemented with insulin (10 μg/ml) and gentamicin (100 μg/ml; 2× medium). One milliliter of the mixture was layered onto 35-mm dishes to create a 1% agarose base. Liquefied 0.6% agarose was mixed with an equal volume of 2× medium, and 10 ml of this solution were mixed with 1 mL of growth medium containing 1×105 ACCM-ER1 cells to yield 1×104 cells/mL in 0.27% agarose; 1 ml of this cell suspension was layered on top of the 1% agarose base, and 1 ml of DMEM/F12 containing 5% FBS was added. The cells were incubated for 2 weeks in the presence of E2 (10 nM). Counts were performed according to the size of the colonies.
Cell migration assay. Cells were seeded in 6-well plates at a density of 1.0×105/well. The following day, the bottom of each well was scratched with a pipette tip. Wells were rinsed with medium to remove detached cells and replaced with serum-free medium. E2 was added to the cultures for 24 h. Images of each well were acquired immediately following wound generation and again after 24 h. Image J software (National Institutes of Health, Bethesda, MD, USA) was used to measure areas that were free of migrating cells. Experiments were performed in triplicate.
Statistical analysis. Statistical comparisons were made using the Student's t-test as appropriate. p-Values <0.05 were considered statistically significant. All statistical tests were performed using Statcel2 software (Statcel2, OMS, Tokyo).
Results
ER transfection in ACCM cells. ACCM cells were transfected with a vector encoding ER. A population of transfected cells was screened by western blotting (Figure 1). We identified three ACCM-ER cell lines (ACCM-ER1–3) that have various levels of ER expression and analyzed them by western blotting. T47D cells expressing human ER were used as positive controls in this transfection. Hence, the ACC-ER1, which has abundant and same expression levels as T47D cells from the breast, was used for further experiments.
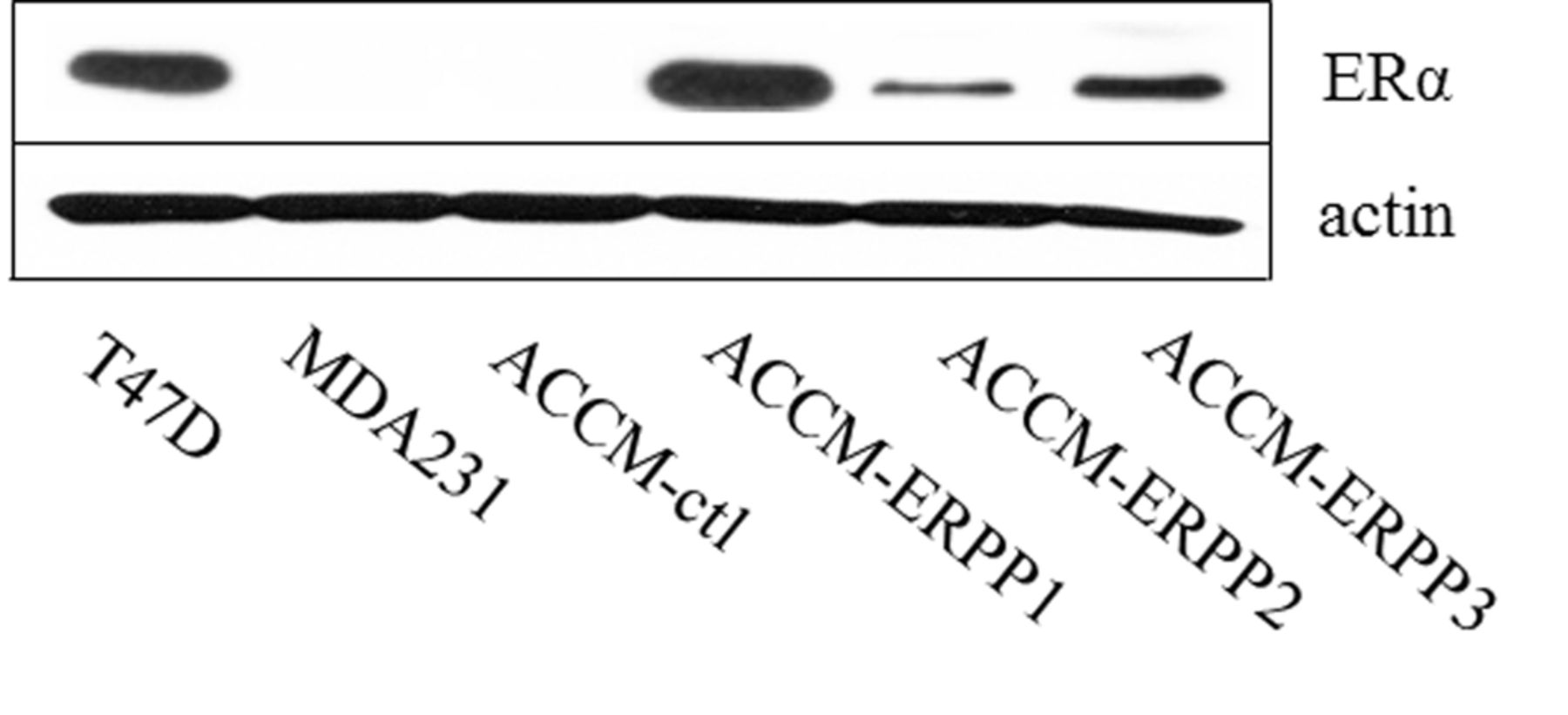
ER expression in ER-deficient ACCM-ER cells. Results were pooled from three cell populations (ACCM-ER1–3) expressing various levels of ER protein, as determined by western blotting. Actin served as a loading control. ACC-ER1, whose abundance was the same as in T47D cells from breast, was used for further experiments.
Effect of E2 on cell cycle in ACCM-ER1 cells. A flow cytometry analysis revealed that the size of the S-phase fraction was increased in a dose-dependent manner by E2 application and almost reached plateau at the concentration of 1 nM (Figure 2). From the result, the concentration of E2 were 10 nM for subsequent experiments.
Effect of E2 on cell proliferation of ACCM-ER1 cells. Treatment with 10 nM E2 for 24 h enhanced the proliferation of ACCM-ER1 cells relative to control (Figure 3A). There was a significant difference between control cells and ER transfectants (p<0.01).
E2 enhances telomerase activity of ACCM-ER1 cells. After the 10 nM of E2 treatment for 24 h Telomerase activity increased in ACCM-ER1 cells. Expression of hTERT, the catalytic subunit of telomerase that serves as a marker for telomerase activity, was also up-regulated in the presence of E2, as determined by RT-PCR (Figure 3B).
E2 regulates ability of ACCM-ER1 cells to form colonies and cell migration. We used colony formation and scratch assays to examine the effects of E2 application on additional aspects of ACCM cell aggressiveness. The application of E2 induced a significant increase in the number of colonies when we used the colony formation assay in ACCM (Figure 4A; p<0.05), as well as cell migration in the scratch assay after 24 h, when compared to control cells (Figure 4B; p<0.05). Cell migration was markedly enhanced in the presence of E2, with the entire scratch area being filled after just 24 h in contrast to control cells. NIH Image (National Institutes of Health, Bethesda, MD) was used to measure the areas not covered by the migrating cells. These experiments were performed in triplicate.
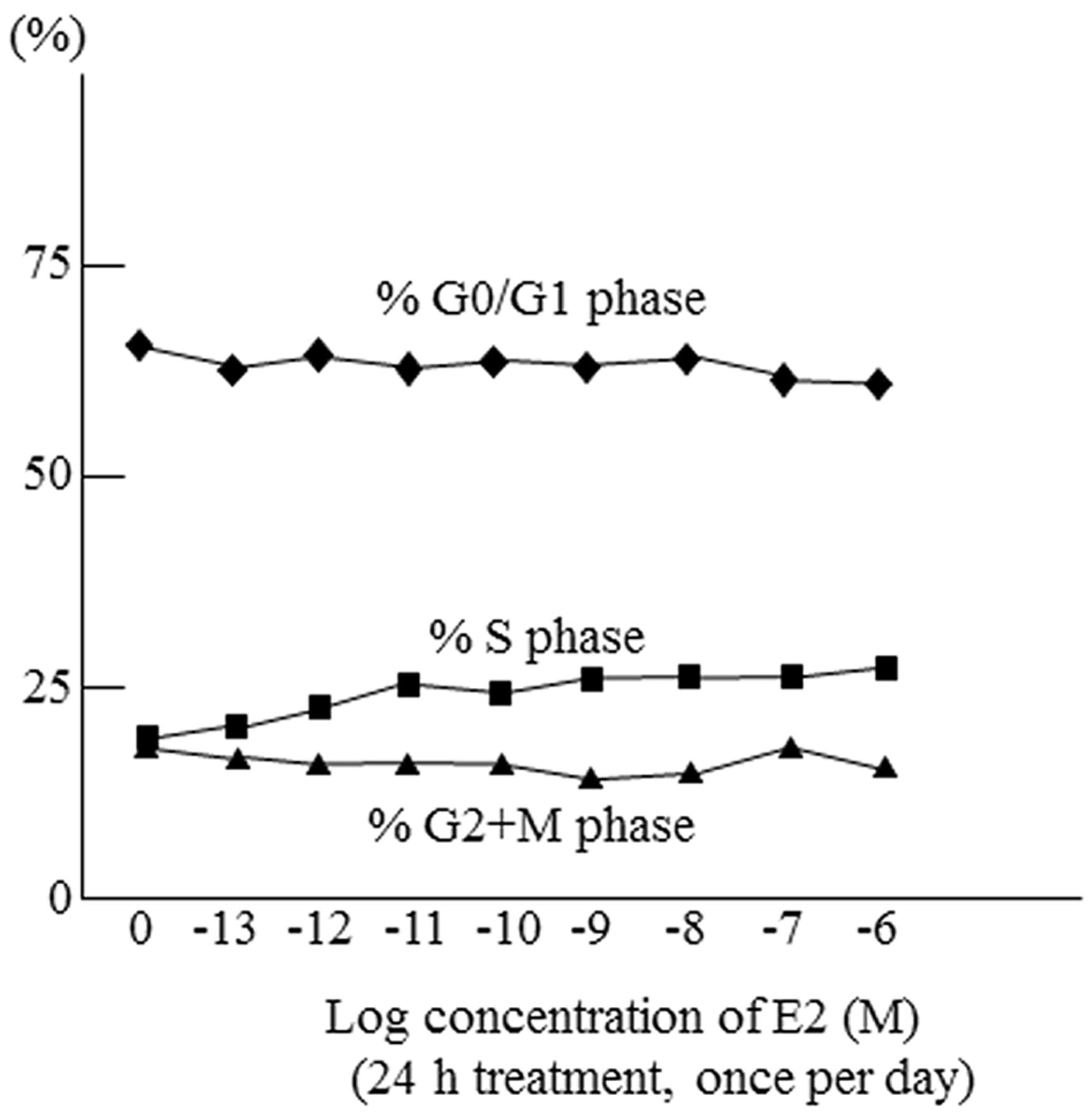
Effect of E2 on ACCM-ER1 cell-cycle distribution. The S phase fraction was increased in a dose-dependent manner by E2 treatment, as determined by flow cytometry.

Effect of E2 treatment on ACCM-ER cell proliferation. (A) ACCM cell proliferation was enhanced in the presence of E2. Treatment with 10 nM E2 for 24 h enhanced the proliferation of ACCM-ER1 cells relative to control cells. (B) Telomerase activity was increased and hTERT transcript level was up-regulated in the presence of E2.

Effect of E2 on ACCM-ER1 for anchorage-dependent growth and cell migration. (A) The application of E2 triggered a significant increase in the number of colonies. (B) ACCM-ER1 cell migration was stimulated by E2 treatment; cells completely covered the scratch area after 12 h, in contrast to control (ctl) cells.
Discussion
The expression levels of steroid hormone receptors is a strong prognostic factor in patients with breast cancer and have been used in clinical management as an indicator of endocrine responsiveness (15, 23). Many advanced breast cancers are negative for PR and ERs and fail to respond to endocrine therapy (24, 25).
SGC is difficult to treat due to ineffectiveness of chemo- and radiotherapy. Although surgical resection is the ideal treatment, it is complicated by histological variability and the slow growth of SGC. However, it was reported that about 66.7% of high-grade SGCs was positive for ER (26). Therefore, the next target was decided to be ER. ER has two sub-types ERα and ERβ, and ERβ was considered to be a potent tumor suppressor in SGC and breast cancer (27, 28, 29).
We previously reported that malignant phenotypes were drastically reduced in PgR-negative breast cancer cells by re-introduction of PgR (9). A similar effect was observed in SGC (13). In the present study, we examined whether ERα expression would have the same effect in SGC. The cell line derived from SGC did not express ERβ (data not shown). However, our results were opposite to those observed by expressing PgR in ACCM cells (13). Three cell lines were established from the human salivary gland, and these exhibited an aggressive character, which may be an important requirement for establishing these cell lines. In fact, ACCM cells were negative for ER, PgR, and androgen receptor. However, other studies have reported the expression of varying ratios of these sex steroid hormones in clinical samples (20, 26, 31). Positive immunoreactivity depends on the specificity of the antibody used; it is therefore difficult to compare our results with those of previous studies. However, the EP1 clone that is the antibody against ER has been already checked regarding its specificity (32), therefore this antibody was used in this study. The detection of sex steroid hormone receptor expression in clinical samples suggests that SGC that is positive for hormone receptor expression has a possibility to respond to hormone therapy. Moreover, ACC grows slowly relative to other malignancies. Therefore, strategies that cause tumor dormancy may also be effective, although not all ACC cells would be destroyed. Recently, it has been reported that some types of SGC maybe related to androgen-regulated protein (33). These findings also show the potential of hormonal therapy for SGCs.
In this study, cell proliferation and migration is induced by E2 application. Moreover, anchorage-dependent growth was examined. A more detailed analysis of the mechanistic basis for the effects of hormones on SGC cell proliferation and migration is required. In a preliminary study, we found that p21, c-myc, and ID1 levels were altered by E2 treatment; future studies will further assess the role of these molecules.
Interestingly, androgen receptor is up-regulated in salivary ductal carcinoma (34-37), which may be linked to tumor progression and patient prognosis. Salivary ductal carcinoma is highly aggressive and is associated with poor prognosis; therefore, the role of androgen and its receptor in SGC warrants closer examination. Moreover, human epidermal growth factor receptor 2, tyrosine kinase and angiogenesis inhibitors, and antibodies have been suggested to be effective for SGC treatment (38). In terms of estrogen axis, clarifying the molecular basis of their effects can optimize the treatment of ERα-positive ACCs. The similar therapeutic strategy may be able to be applied. We have to perform the experiments treated with tamoxifen not only in vitro but also in vivo.
In conclusion, the results of this study indicate that the E2-ER system may be a potential strategy for treatment of ER-positive SGC.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 15K11257. The authors gratefully acknowledge this financial support. The Authors would like to thank Editage (www.editage.jp) for English language editing.
Footnotes
Conflicts of Interest
None of the Authors has a financial conflict of interest to disclose in relation to the content of this article.
- Received April 5, 2016.
- Revision received May 11, 2016.
- Accepted May 17, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.