


Abstract
Papillomaviruses induce a range of benign and malignant lesions in their hosts, including cervical cancer, that is associated with high-risk human papillomavirus (HPV) types. The nuclear factor kappa-light-chain-enhancer of activated B-cells (NFκB) plays a pivotal role in HPV-infected cells, and its expression and activity are modulated by several viral oncoproteins. NFκB modulation seems to first facilitate viral persistence and immune evasion, and later to drive tumour progression, but the many conflicting results and the complexity of its signaling networks require great prudence while interpreting the role of NFκB in papillomaviral lesions. Accordingly, the pharmacological targeting of the NFκB pathway in HPV-induced lesions is a complex and currently unmet challenge. This review deals with recent findings concerning NFκB activation in HPV-infected cells, its role in viral persistence, cell transformation and tumour progression, and with current efforts to target this pathway for cancer prevention and therapy.
Human papillomaviruses (HPV) are associated with a variety of benign and malignant lesions. In particular, HPV types 16 (HPV16) and 18 (HPV18), have long been recognized as important biological carcinogens, and are particularly associated with cervical cancer and other anogenital malignancies (1). Despite recent advances in cancer prevention, cervical cancer remains a major public health problem, with an estimated worldwide incidence of 527,624 new cases in 2012 (2). For 2013, 12,340 new cervical cancer cases were expected in the United States, as well as 4,030 deaths (3). Although HPV vaccination is expected to considerably reduce the burden of cervical cancer in developed countries, its implementation is slow in the developing world, maintaining high disease burdens (2). Another reason for concern is the increasing worldwide incidence of non-genital HPV-associated cancer, particularly HPV-associated oropharyngeal cancer (4). High-risk HPVs encode several oncogenes, most notably E6 and E7, which promote the neoplastic transformation of infected cells (5). The viral oncoprotein E6 leads to the ubiquitination and subsequent proteasomal degradation of p53, while E7 blocks the function of retinoblastoma protein (pRb), deregulating the cell cycle and blocking DNA-repair mechanisms and apoptosis.
Advanced cervical cancer poses a particularly difficult therapeutic challenge. In particular, there is a clear need for improvement in therapy for recurrent or metastatic cancer, associated with short survival periods. The standard treatment for metastatic and recurrent cervical cancer has long been cisplatin, as monotherapy or combined with radiotherapy, with a median survival of 6 months (6). However, cancer cells develop cisplatin resistance, requiring combination therapies with paclitaxel, topotecan, gemcitabine, vinorelbine or ifosfamide, in order to achieve improved median survival of 10 to 12.9 months. Monoclonal antibodies and inhibitors of receptor tyrosine kinases targeting vascular endothelial growth factor receptor (VEGFR), stem-cell growth factor receptor (c-KIT) and platelet-derived growth factor receptor (PDGFR) are also under study in clinical trials (6).
In many types of cancers, including cervical cancer, resistance to chemotherapy has been found to be strongly dependent on the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) (7-12). The NFκB pathway and its regulation are highly complex, and may lead to cancer invasion and chemoresistance in multiple ways, including resistance to apoptosis, metabolic changes and modulation of the tumoural microenvironment through the production of inflammatory mediators (13).
This review deals with recent findings concerning NFκB activation in HPV-infected cells, its role in viral persistence, cell transformation and tumour progression, and with current efforts to target this pathway for cancer prevention and therapy.
The NFκB Pathway
The NFκB transcription factor is composed of homo- or heterodimers formed by five proteins belonging to the avian reticuloendotheliosis viral oncogene homolog (REL) which share a DNA-binding domain and a dimerization domain, the REL homology domain. These REL proteins include p50 (NFκB1), p52 (NFκB2), p65 (RELA), RELB and c-REL (REL), as reviewed by Perkins et al. (13). p50 and p52 are translated in the form of two protein precursors, known as p105 and p100. Proteasomal processing of these two proteins produces p50 and p52 (14). Binding of NFκB to DNA leads to transcription of its target genes, thereby modulating numerous cell functions.
Cells can rapidly activate NFκB signaling as a first line of defence against infection or otherwise stressful conditions. This requires most REL proteins to be pre-synthesized and kept in the cytoplasm in an inactive form, ready to be activated in response to adequate stimuli. REL proteins are bound by and sequestered in the cytoplasm by a family of inhibitors of NFκB (IκBs) proteins which include IκBα, IκBβ and IκBε (15). The ankyrin repeat motifs found in IκB proteins allow them to bind the REL subunits and are also present in the C-termini of p100 and p105, which also act as IκB-like NFκB inhibitors (16). IκB proteins also have other functions (17), although these are outside the scope of the present review.
The NFκB pathway is highly complex and subject to multiple regulatory factors that help shape its signalling activity to each cell's conditions. Figure 1 depicts certain essential aspects of the NFκB pathway, including its canonical and non-canonical activation routes. As mentioned above, the canonical pathway provides a rapid responds to stressful stimuli by activating pre-existing REL proteins. The non-canonical pathway is involved in lymphoid organogenesis, B-cell maturation and survival, and bone metabolism, and requires the production of p52 from its inactive precursor p100, as reviewed by Sun (16). In the canonical pathway, the binding of a ligand to a receptor on the cell membrane, such as a tumour necrosis factor (TNF) receptor or a member of the interleukin-1 receptor/toll-like receptor (TLR) superfamily, allows the recruitment of adaptor proteins such as TNF receptor-associated proteins (TRAFs).
TRAF proteins recruit and activate the IκB kinases alpha (IKKα) and beta (IKKβ), which together with a scaffold protein known as NFκB essential modulator (NEMO or IKKγ), form the IKK complex (18). The activated IKK complex phosphorylates two serine residues (ser23/ser36) in the IκBα regulatory domain, targeting it for ubiquitination and proteasomal degradation (15). IκB phosphorylation allows the release of REL proteins and their translocation into the cell nucleus, where they may bind DNA. The non-canonical pathway is activated by non-inflammatory stimuli, such as the binding of B cell-activating factor (BAFF) to its receptor (BAFFR) (16). The non-canonical pathway is critically mediated by NFκB-inducing kinase (NIK) which activates IKKα (but not IKKβ or IKKγ), leading to p100 phosphorylation, ubiquitination and consequent proteasomal processing to generate the active p52. Processing of p100 also abolishes its IκB-like inhibitory activity. The RELB/p52 dimer is then translocated to the nucleus, where it exerts its activity as a transcription factor. NFκB signaling may also proceed through atypical pathways initiated in response to DNA damage, which provide the link between genotoxic anticancer agents and NFκB activation (19). Poly(ADP-ribose) polymerase 1 (PARP1) senses DNA damage and mediates phosphorylation of the NFκB essential modulator (NEMO) and its binding to small ubiquitin-like modifier (SUMO, SUMOylation) through the ataxia telangiectasia mutated (ATM) kinase, ultimately leading to NFκB activation.
HPV-Induced Carcinogenesis
High-risk HPVs, most commonly HPV-16 and HPV-18, are the aetiological agents of most anogenital (20, 21) and of a significant number of oropharyngeal (4, 22) carcinomas. The involvement of HPV in malignancies such as oesophageal cancer remains a matter of debate. Other papillomavirus induce a range of benign and malignant lesions in animal species (23). As previously mentioned, the genome of high-risk HPVs encodes a number of oncogenes, expressed early after viral infection. These early genes are located in the so-called early region of the viral genome, as opposed to the late region, encoding structural proteins. Additionally, the HPV genome also contains regulatory elements, namely the long control region (LCR), involved in regulating gene expression (24). HPV oncogenes, most notably the E6 and E7 genes, promote the proliferation of infected cells, while blocking DNA repair and apoptosis, thus potentially leading to the accumulation of genetic mutations and to carcinogenesis, as reviewed by Klingelhutz and Roman (25). In fact, the E6 protein interacts with the critical tumour-suppressor p53, efficiently promoting its ubiquitination and subsequent proteasomal degradation (26). p53 protein is normally present at low levels, which are raised in response to genotoxic or cytotoxic stress. Up-regulation and activation of p53 activates multiple pathways leading to DNA repair, cell-cycle arrest and apoptosis. Apart from its central role in inactivating p53, the E6 oncoprotein also contributes to tumourigenesis by activating telomerase, disrupting epithelial-cell adhesion, polarity and differentiation, altering gene transcription and reducing immune recognition of infected cells, as reviewed by Howie et al. (27). The E7 protein binds to and induces degradation of pRb, thus deregulating the cell cycle and allowing suprabasal cells to maintain unchecked proliferative activity (28, 29).
Initially, HPVs invade mitotically active basal cells of stratified squamous epithelia, presumably through microwounds, using specific cell-surface receptors (30). Viral genomes are produced at 50-100 copies per cell, maintained in episomal form and segregated to daughter cells with support from the E1 and E2 early viral proteins. As cells migrate to suprabasal epithelial strata, they differentiate but are kept in a proliferative state by the E6 and E7 oncoproteins. This allows the expression of the late genes L1 and L2, encoding the capsid proteins, which is dependent on squamous cell differentiation, and the replication of viral DNA, culminating in virion assembly and release with mature squamous cells (5). Integration of viral DNA into host chromosomes is a comparatively infrequent but critical event, as it leads to E6 and E7 overexpression, and is believed to be the fundamental event for HPV-induced cell transformation (31). HPV-driven carcinogenesis is a well-characterized multistep process that typically develops from pre-malignant intraepithelial hyperplastic and dysplastic lesions, through in situ carcinoma, to invasive squamous cell carcinoma. Advanced and metastatic cancer poses significant therapeutic challenges due to the development of chemoresistance, a trait that is often dependent on NFκB signaling (12).
NFκB Modulation by HPV Oncoproteins
Early studies suggested that high-risk HPV oncoproteins modulate the expression of NFκB-responsive genes, pointing towards their intervention in NFκB activation (32-34). James et al. reported that HPV16 E6 activates NFκB and induces the expression of its anti-apoptotic target, inhibitor of apoptosis C2 (cIAP2) (35). The authors also concluded that this effect depended on the presence of the interaction domain named PSD95/Dlg/ZO1 (PDZ) binding motif of the E6 protein, which mediates its interaction with other PDZ-containing proteins. Activation of NFκB p52-containing complexes, presumably activated through the non-canonical pathway, was demonstrated, resulting in cIAP2 up-regulation and abrogation of TNF-induced apoptosis. Importantly, cIAP2 is considered a critical antiapoptotic factor in cells expressing HPV16 oncoproteins and its knockdown is sufficient to induce apoptosis in HeLa cells or in HPV16-immortalized human oral keratinocytes (36). Taken together, these findings suggest that NFκB is activated by E6 and that abrogation of NFκB signaling is an interesting therapeutic target in HPV-induced cancer.
Additional details concerning the mode of NFκB activation by E6 were provided by later studies (Figure 2). Under hypoxic conditions, the E6 oncoprotein of high-risk HPVs was found to stimulate the ubiquitination and proteasomal degradation of cylindromatosis (CYLD) lysine 63 deubiquitinase, a negative regulator of the NFκB pathway that blocks TRAF-mediated IKK recruitment and activation (37). CYLD acts to deubiquitinate TRAF2, TRAF6 and NEMO, and CYLD inactivation up-regulates NFκB signalling in a sustained way, both in vitro and in vivo, under hypoxic conditions, such as those commonly observed in cervical or head-and-neck cancer. These findings are in accordance with the established link between hypoxia, aggressive biological behaviour and chemoresistance. Two years later, Xu et al. suggested that the E6-mediated activation of NFκB might also be mediated by the inactivation of the nuclear transcription factor X-box binding 1 (NFX1), which acts to inhibit NFκB (38). The authors also showed that NFX1 up-regulates the expression of p105, which acts to inhibit NFκB before it is cleaved to generate active p50. As E6 expression also resulted in p105 down-regulation and NFκB activation, the authors suggested this occurs through inhibition of NFX1-mediated p105 expression.
Other oncoproteins were suggested to be implicated in NFκB activation by HPV. Kim et al. reported that the HPV16 E5 oncoprotein up-regulated cyclo-oxygenase-2 (COX2) by activating NFκB (and, to a lesser extent, activator protein-1, AP1) signalling (39). The E2 protein of α, β, and μ-HPV types was shown to enhance the activation of NFκB stimulated by TNF but not by IL1 (40). This was mediated by direct TRAF5 binding and activation and was independent of the NFκB regulator TAX1 binding protein-1 (TAX1BP1). The authors speculated that this mechanism might be involved in the differentiation of infected keratinocytes, allowing the implementation of the productive viral cycle. These findings are particularly relevant as they point towards a general mechanism common to many HPV types and also because they suggest that NFκB activation may be an early event after HPV infection and not necessarily confined to advanced lesions. Hussain et al. demonstrated that both the E6 and E7 proteins from cutaneous HPV38 activate the canonical (but not the non-canonical) NFκB signalling pathway, up-regulating the expression of anti-apoptotic genes cIAP1, cIAP2 and xIAP, and effectively inhibiting TNF or UV-mediated apoptosis (41).
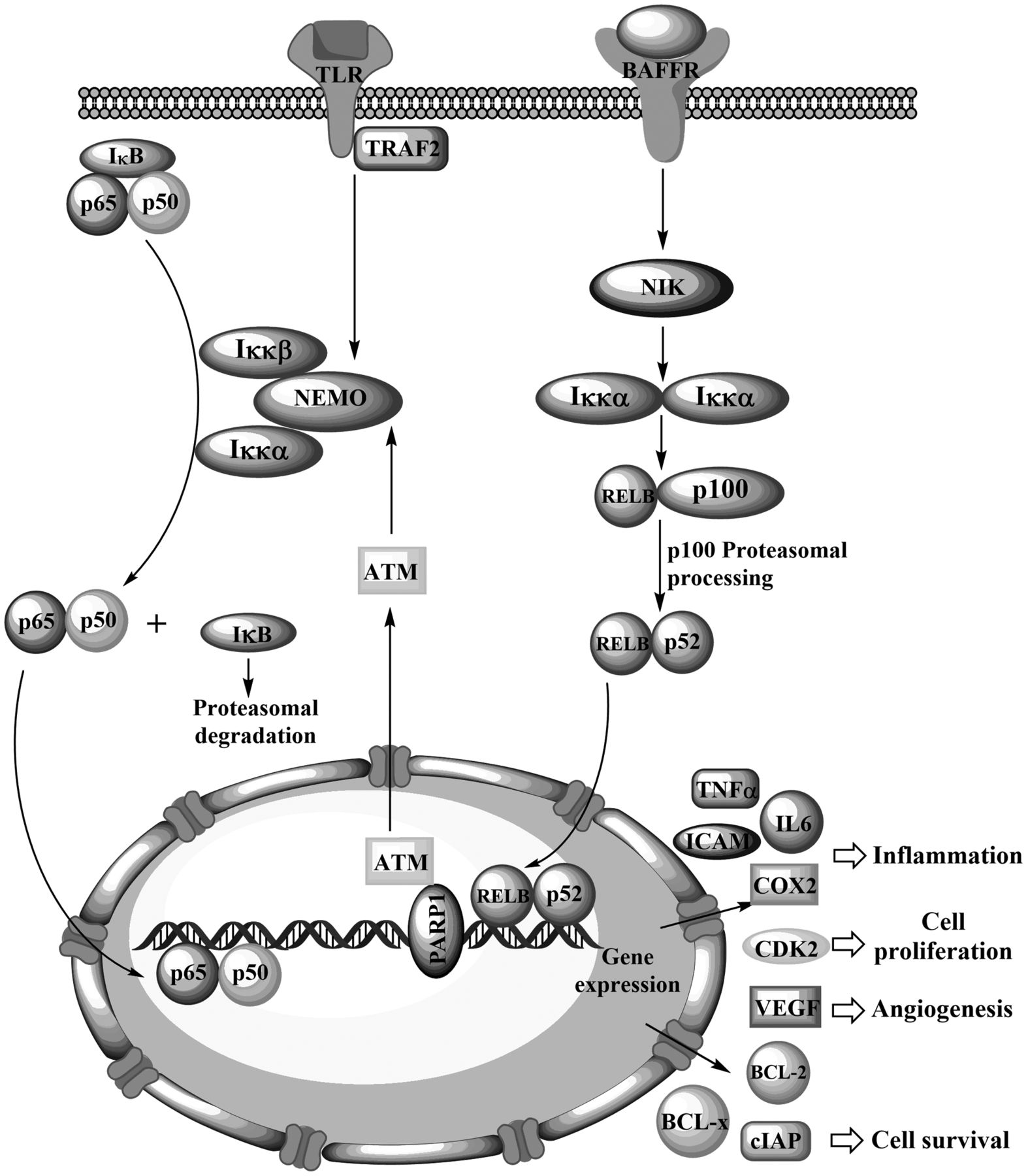
An overview of the canonical and non-canonical pathways leading to nuclear factor kappa-light-chain-enhancer of activated B-cells (NFκB) activation.
The close relationship between NFκB and cancer was suggested by early studies showing that the turkey retrovirus REV-T encodes the v-rel oncogene, a homologue of the NFκB DNA-binding subunits (42). Numerous epithelial and lymphoid malignancies show NFκB activation, either due to mutations leading to high NFκB signalling, or driven by continuous NFκB stimulation (e.g. by cytokines produced by tumour-associated macrophages), or even in response to common chemotherapeutic agents, such as cisplatin (13, 43-46).
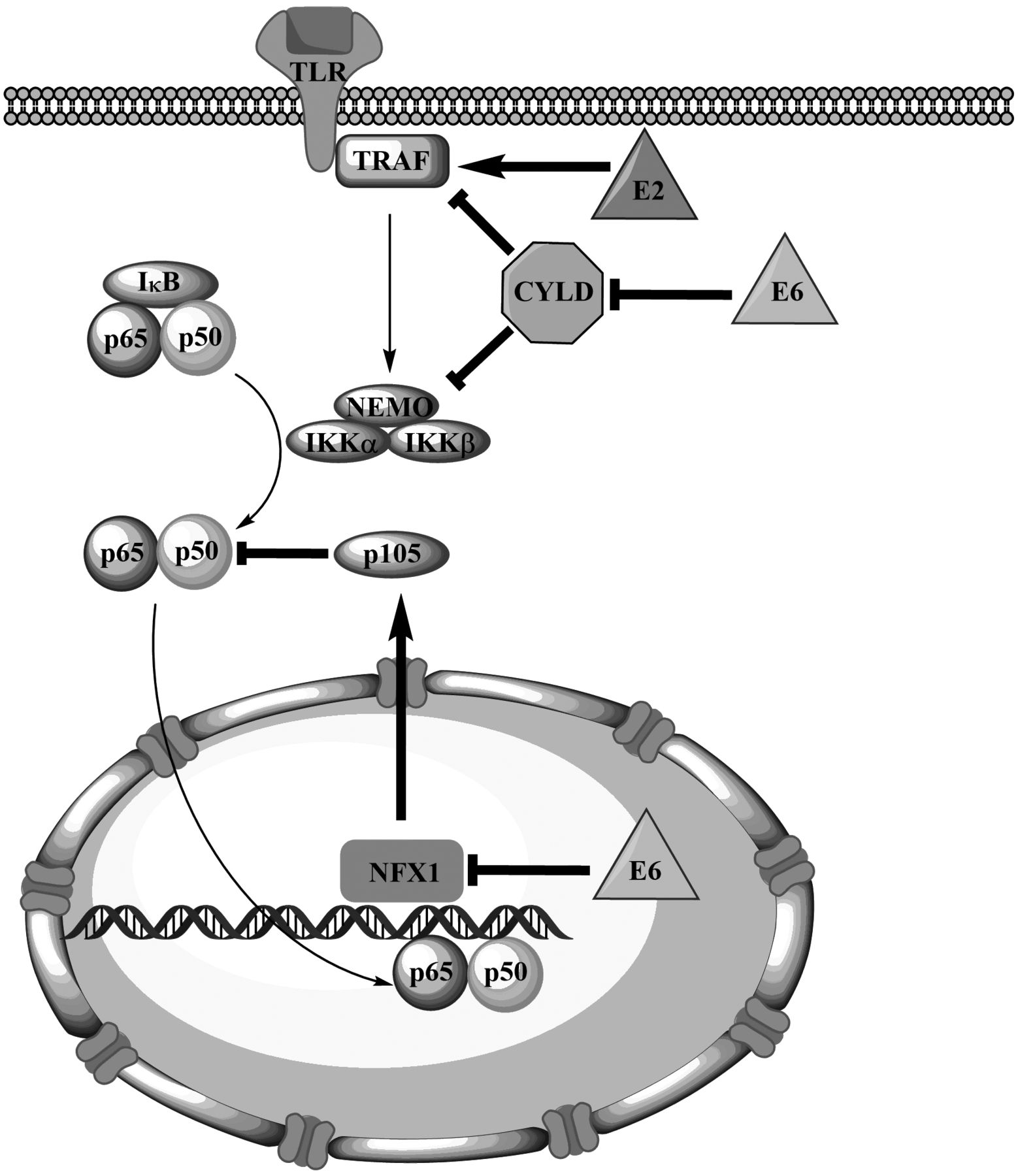
Activation of the canonical nuclear factor kappa-light-chain-enhancer of activated B-cells (NFκB) pathway by human papillomavirus (HPV) oncoproteins E2 and E6. Normal arrows depict the normal signaling pathway, while arrows in bold depict the action of HPV oncoproteins (straight arrows indicate activation, blunt arrows indicate inhibition).
NFκB activation regulates the transcription of a number of diverse genes encoding proteins and miRNAs, thereby regulating inflammation, cell survival, proliferation, energy metabolism and adhesion, as well as the cellular microenvironment (47, 48). However, the effects of NFκB signaling depend on the cell type involved: neoplastic cells often exhibit a ‘malignant’ version of the protective role played by NFκB in normal cells (13). In fact, NFκB activity closely recalls all the hallmarks of cancer (49). Among the many genes transcriptionally influenced by NFκB, some encode proteins involved in inflammation (e.g. COX2, TNF, IL6, and intercellular adhesion molecule, (ICAM)), cell survival (cIAP, XIAP, B-cell lymphoma-2 (BCL2) and BCL-x), proliferation (cyclin-dependent kinase 2, CDK2), and angiogenesis (VEGF). This central position of NFκB, between inflammatory, proliferative and survival pathways, marks it as a key player in the link between inflammation and cancer. NFκB can also promote the metabolic switch from oxidative phosphorylation to glycolysis even in the presence of oxygen, known as the Warburg effect, as reviewed by Tornatore et al. (50). In the presence of functional p53 protein (e.g. in normal cells) p65 interacts with p53. Both proteins enter the nucleus to up-regulate the expression of metabolic genes which encode, for instance, the complex IV subunit of cytochrome c oxidase, increasing aerobic metabolism and reducing glycolysis (51). In parallel, p53 also down-regulates expression of the glucose receptor GLUT3. However, in the absence of p53 (e.g. in p53−/− or HPV-induced cancer), p65 interacts with mortalin which mediates its translocation into the mitochondria, suppressing mitochondrial function (52). Simultaneously, NFκB up-regulates GLUT3 expression, thus elevating intracellular glucose levels and favouring glycolysis. This is an example of how the outcome of NFκB signalling depends on the cellular context, in particular on the status of tumour suppressors such as p53 and phosphatase and tensin homolog (PTEN). The inactivation of tumour suppressors can drive NFκB signalling towards an oncogenic, tumour-promoting outcome (13). This is of particular significance in HPV-induced tumours, which depend on p53 degradation by E6 and commonly bear wild-type p53. Due to the complexity of the NFκB pathway, it is even possible that NFκB activation may result in a pro-apoptotic affect, depending on several factors such as cell type, the nature of the stimulus, presence of tumour suppressors, and histone acetylation status, as reviewed by Perkins (13) and Godwin et al. (53). For instance, NFκB was shown to mediate apoptosis triggered by wild-type p53 in vitro (54). It was also demonstrated that NFκB can activate p53 and target polo-like kinase 3 (PIK3) to phosphorylate p53, thus increasing its half-life and potency (55).
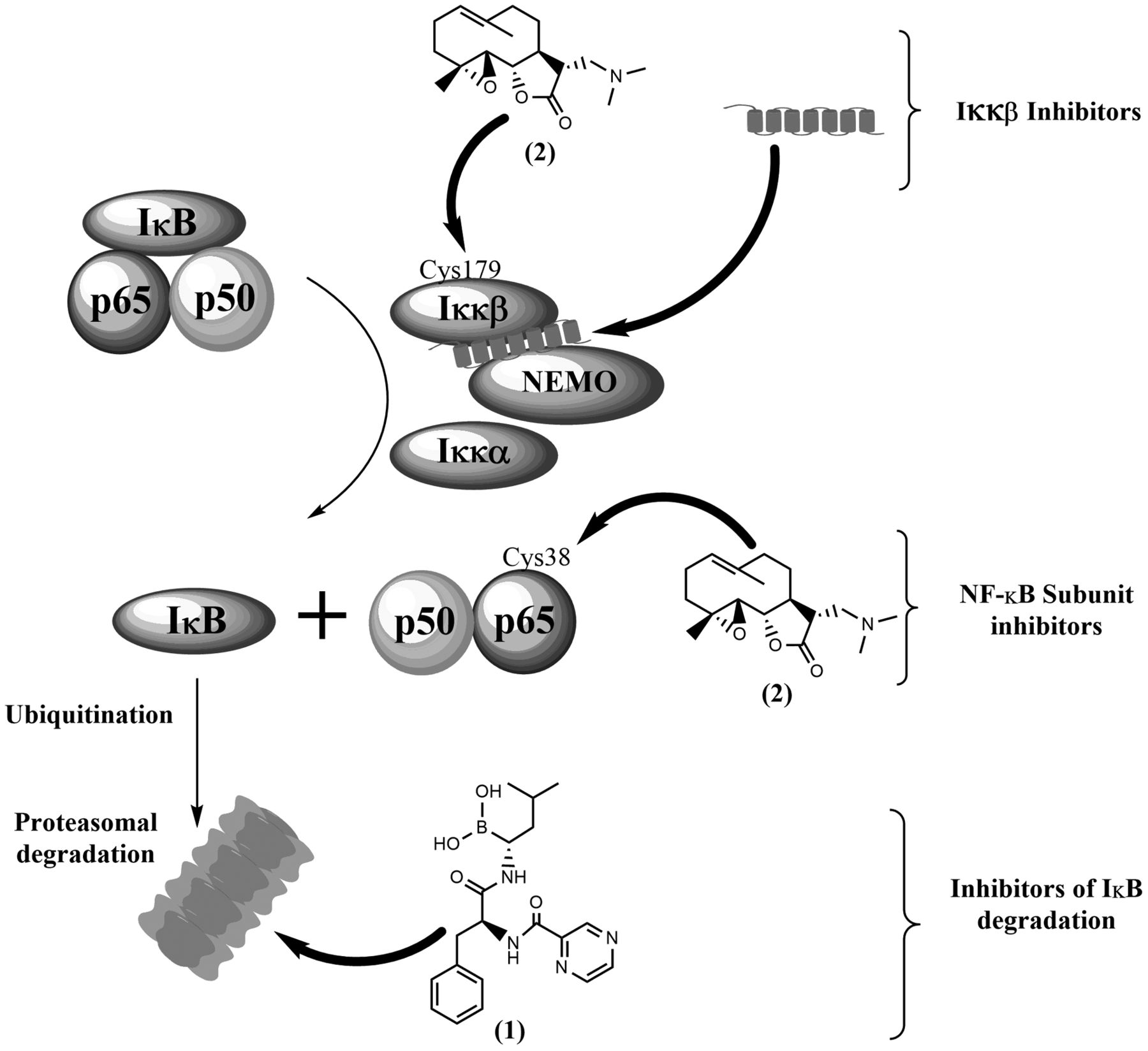
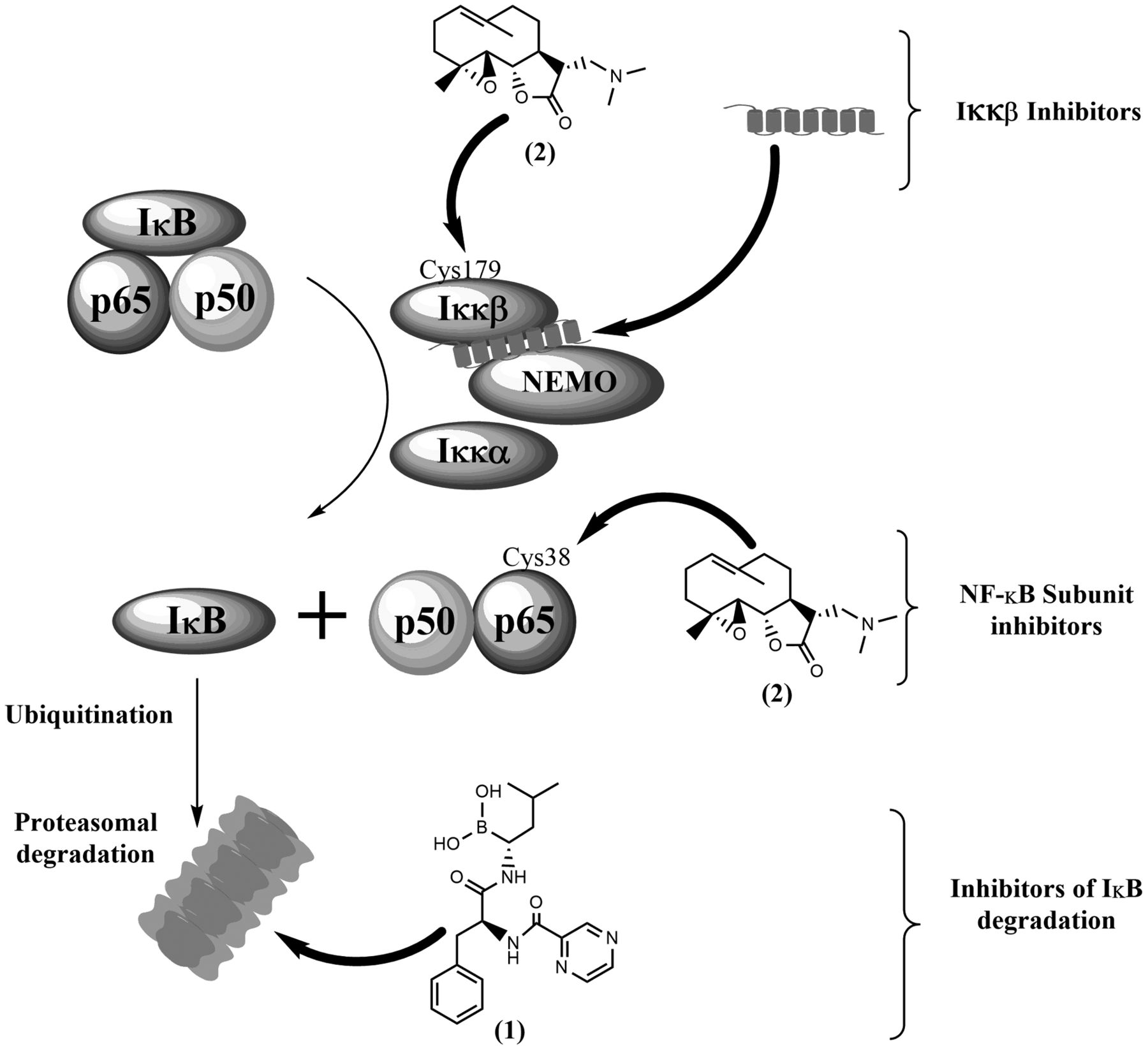
Some mechanisms of nuclear factor kappa-light-chain-enhancer of activated B-cells (NFκB) inhibition, including: inhibition of inhibitor of NFκB (IκB) degradation by proteasomal inhibitor bortezomib (1), IκB kinase (IKKβ) inhibition by covalent binding of dimethylaminoparthenolide (2) to Cys179 or interaction of an inhibitory peptide with the NFκB essential modulator (NEMO)-binding domain, and direct inhibition of p65 activity by covalent binding of dimethylaminoparthenolide (2) to Cys38. Normal arrows depict the normal signaling pathway, while arrows in bold depict the action of NFκB inhibitors.
A well-established line of evidence shows that some types of cancer induced by high-risk HPV display NFκB activation. NFκB was reported to be constitutively activated in cervical cancer and intraepithelial lesions (56, 57). NFκB DNA-binding activity progressively increased from low-grade towards high-grade lesions and invasive cancer. Interestingly, the formation of p50–p50 homodimers, rather than the common p50–p65 heterodimers was reported. Later, Li et al. described a statistically significant association between NFκB activation and tumour progression, aggressive biological behaviour (higher histological grade, lymphatic metastasis, interstitial invasion and larger tumour size) and poor prognosis (lower survival rates) in cervical cancer, suggesting NFκB as a potential therapeutic target (58). Constitutive NFκB activation was also reported for HPV-positive oral cancer, showing p50–p65 heterodimerization (59). However, a recent study suggests that NFκB activation is more frequently observed in HPV-negative than in HPV-positive head-and-neck cancers (60). While there seems to be a clear role for NFκB in cervical cancer, the case for HPV-positive head-and-neck cancer requires additional efforts before any definite conclusions may be drawn.
A second line of evidence shows that NFκB signaling triggers important protective antiviral responses and that under certain conditions, viral oncoproteins inactivate NFκB, averting such responses (61). Early this century, Spitkovsky et al. showed that E7 associates with and represses the IκB complex, effectively down-regulating NFκB (62). Signalling by NFκB is essential in order to activate dendritic cells and trigger protective immune responses (63). High-risk HPV types were found to impair the innate immune response usually triggered by infected keratinocytes, by up-regulating ubiquitin carboxyl-terminal hydrolase L1 and down-regulating NEMO (61). Recently, Nakahara et al. showed that HPV16 E1 protein activates NFκB, triggering a feedback loop which limits viral replication (64). These findings suggest that circumventing NFκB activation may be a valid strategy to promote viral replication and persistence. This line of evidence draws attention to the protective role of NFκB signalling during the early phases of viral infection, and its importance for establishing a protective immune response, able to eradicate HPV infection, preventing viral persistence and potential cell transformation.
Targeting NFκB
In 2006, over 750 NFκB inhibitors are known, including antioxidants, small molecules, peptides, small RNA/DNA, microbial and viral proteins (65). The present review focuses on general NFκB targeting mechanisms, while presenting some of the most promising molecules, in the context of HPV-induced cancer. The rationale underlying NFκB inhibition for treating cancer is clear: inhibiting the anti-apoptotic and cancer-promoting functions of NFκB will counteract the aggressive biological behaviour of cancer and, if included in combination therapies, will sensitize them to conventional drugs. However, the complexity of the NFκB pathway and its regulatory mechanisms – discussed below – make it necessary to carefully design appropriate targeting strategies in order to avoid off-target effects and associated toxicities. Inhibitors of the NFκB pathway are numerous and variably specific, but can be grouped into those targeting IKK activation, IκB degradation and NFκB DNA-binding (Figure 3), as proposed by Nakanishi and Toi (66).
Several highly specific small-molecule IKKβ inhibitors have been developed which have interesting anti-inflammatory and anti-neoplastic activity (67-70). In general, these compounds act as ATP analogues with some degree of specificity for IKKs, molecules with allosteric effects on IKK structure, and compounds that interact with Cys179 in the IKKβ activation loop (65). However, inhibiting IKKs is likely to have severe physiological implications, e.g. in the regulation of inflammatory processes, and such drugs may exert significant off-target effects in vivo (71,72). A cell-permeable peptide spanning the IKKβ NEMO-binding domain was found to disrupt TNFα-induced NFκB activation in vivo, preventing inflammation in animal models (73). Interestingly, this peptide was reported to allow the maintenance of basal IKK activity while suppressing enhanced activation by pro-inflammatory cytokines, which may help minimizing the side-effects associated with more drastic NFκB inhibition.
Inhibiting IκB degradation by blocking the proteasome has been a successful strategy in treating certain haematopoietic malignancies. Bortezomib (1 in Figure 3), a proteasome inhibitor, has been approved for clinical use in newly diagnosed and relapsed/refractory multiple myeloma and multiple mantle cell lymphoma. However, proteasome inhibitors are expected to have other effects besides inhibiting NFκB, and significant toxicity has been reported with bortezomib use, namely peripheral neuropathy (74). Despite its success with haematopoietic malignancies, single-agent or combination therapies including bortezomib have shown only limited efficacy against solid tumours such as melanoma (phase II clinical trial NCT00512798), metastatic breast cancer (75), and urothelial cancer (76), as reviewed by Cao et al. (77). A second generation of proteasome inhibitors (e.g. marizomib, oprozomib and delanzomib) holds the promise of providing greater efficacy with reduced toxicity, as well as more flexible dosing schedules (78).
Targeting NFκB subunits is a strategy that may provide more specific effects with less toxicity. A peptide spanning the Ser536 p65 phosphorylation site was shown to inhibit NFκB function selectively in response to some inflammatory stimuli in vitro and in vivo (79-80). The activity of NFκB can be inhibited by some natural products, such as sesquiterpene lactones found in Asteraceae plants (81), which possess a reactive α-methylene-γ-lactone group and target a highly conserved cysteine residue in the REL homology domain (e.g. Cys38 in p65) (82, 83). Inhibition of IKKβ by targeting a similar cysteine residue (Cys179) is also thought to play a secondary role in NFκB inhibition by sesquiterpene lactones. Parthenolide is one such NFκB inhibitor, well-known for its anti-inflammatory properties, and has been tested in vitro and in vivo against a range of malignancies, as reviewed by Amorim et al. (84). However, as with many natural products, the pharmacokinetic properties of parthenolide hamper its in vivo efficacy. This problem has been solved through the development of a semi-synthetic aminoderivative, dimethylaminoparthenolide (2 in Figure 3), which provided a favourable pharmacokinetic profile, retaining NFκB-inhibitory properties, and had significant anti-neoplastic activity in vivo, coupled with minimal toxicity (85). In view of these favourable results, dimethylaminoparthenolide is reported to have entered phase I clinical trials against haematological malignancies (86).
When targeting NFκB, it is also important to consider that its outcome is largely determined by interactions with other signalling pathways. As previously mentioned, the cell's p53 status critically determines the effects of NFκB activation. Here again, sesquiterpene lactones have promising characteristics by targeting multiple, potentially synergic pathways. Parthenolide releases p53 from murine double minute 2 (MDM2)-mediated inhibition, thus raising the intracellular levels of active p53, while also generating reactive oxygen species to increase oxidative stress and trigger apoptosis (81). This ability to raise the levels of active p53 in conjunction with NFκB inhibition makes parthenolide and its derivative dimethylaminoparthenolide particularly interesting in the context of HPV-induced cancer, which rely on E6 to degrade wild-type p53. It appears that parthenolide is able to sufficiently up-regulate the levels of functional p53 in the presence of E6, removing one of the fundamental basis for HPV-induced cancer and triggering apoptosis (87).
Curcumin, found in the Indian spice turmeric, is another natural compound, with pleiotropic biological activities, which also acts to inhibit NFκB. Although this is not a targeted therapy, curcumin and other natural anti-inflammatory and antioxidant compounds remain a persistent focus of scientific interest. The chemopreventative and chemotherapeutic effects of these compounds on cervical cancer, largely mediated through inhibition of the NFκB pathway, were recently reviewed (88). In order to obtain effective tissue concentrations, high oral doses of curcumin are required, but gastrointestinal toxicity (fullness, abdominal pain) may be dose-limiting (89). Several strategies have been proposed in order to increase the oral bioavailability of curcumin, including the use of adjuvants, curcumin nanoparticles and analogues and phase II/III clinical trials on patients with cervical cancer are reported to be under way (90).
Conclusion
There exists accumulating evidence on the roles of NFκB in papillomavirus-induced lesions but experimental findings often seem contradictory. This is in line with the complex signaling network involving this transcription factor and the different settings in which it has been studied. In many instances, it seems plausible that NFκB plays a protective role during the early phases of HPV infection and persistence, while promoting tumour progression and resistance to radiotherapy and chemotherapy in advanced lesions. Inhibiting NFκB signaling is a tempting strategy to fight some types of HPV-induced cancer, especially cervical cancer. In fact, some targeted and non-targeted NFκB inhibitors have been successfully tested against HPV induced cancer or cancer cell lines. The development of second-generation proteasome inhibitors, novel formulations for polyphenolic compounds with enhanced bioavailability, and the development of compounds able to simultaneously target both NFκB and p53 makes this a rapidly evolving field of research, likely to contribute with improved therapies for patients with cancer in the near future.
Acknowledgements
This work was financially supported by: Project POCI-01-0145-FEDER-006939 (Laboratory for Process Engineering, Environment, Biotechnology and Energy – LEPABE funded by FEDER funds through COMPETE2020 - Programa Operacional Competitividade e Internacionalização (POCI) – and by national funds through FCT - Fundação para a Ciência e a Tecnologia, and also by European Investment Funds by FEDER/COMPETE/POCI– Operational Competitiveness and Internationalization Programme, under Project POCI-01-0145-FEDER-006958 and National Funds by FCT, under the project UID/AGR/04033/2013. Rui M. Gil da Costa is supported by postdoctoral research grant SFRH/BPD/85462/2012, from FCT, funded by the Portuguese Government and the Social European Fund.
Footnotes
This article is freely accessible online.
Conflicts of Interest
The Authors declare that they have no conflict of interest in regard to this article.
- Received February 7, 2016.
- Revision received April 2, 2016.
- Accepted April 7, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Human Papillomavirus Modulates Matrix Metalloproteinases During Carcinogenesis: Clinical Significance and Role of Viral Oncoproteins
- Systematic analysis of IL-1 cytokine signaling by high-risk HPV oncoproteins
- Expression of LncRNAs in HPV-induced Carcinogenesis and Cancer Cachexia: A Study in K14-HPV16 Mice
- Standardized Uptake Value for 18F-Fluorodeoxyglucose Is a Marker of Inflammatory State and Immune Infiltrate in Cervical Cancer
- Penicillium purpurogenum exerts antitumor effects and ameliorates inflammations in Erlich mice model
- Human Papillomavirus 16 E5 Inhibits Interferon Signaling and Supports Episomal Viral Maintenance
- HPV18 Persistence Impairs Basal and DNA Ligand-Mediated IFN-{beta} and IFN-{lambda}1 Production through Transcriptional Repression of Multiple Downstream Effectors of Pattern Recognition Receptor Signaling
- Curcumin and Rutin Down-regulate Cyclooxygenase-2 and Reduce Tumor-associated Inflammation in HPV16-Transgenic Mice
- Regulation of miRNA-146a and miRNA-150 Levels by Celecoxib in Premalignant Lesions of K14-HPV16 Mice