


Abstract
Background: P21-associated noncoding RNA DNA damage-activated (PANDA) is induced in response to DNA damage and represses apoptosis by inhibiting the function of nuclear transcription factor Y subunit alpha (NF-YA) transcription factor. Herein, we report that PANDA affects regulation of p53 tumor-suppressor protein. Materials and Methods: U2OS cells were transfected with PANDA siRNAs. At 72 h post-transfection, cells were subjected to immunoblotting and quantitative reverse transcription-polymerase chain reaction. Results: Depletion of PANDA was associated with decreased levels of p53 protein, but not p53 mRNA. The stability of p53 protein was markedly reduced by PANDA silencing. Degradation of p53 protein by silencing PANDA was prevented by treatment of MG132, a proteasome inhibitor. Moreover, depletion of PANDA prevented accumulation of p53 protein, as a result of DNA damage, induced by the genotoxic agent etoposide. Conclusion: These results suggest that PANDA stabilizes p53 protein in response to DNA damage, and provide new insight into the regulatory mechanisms of p53.
Recent mass -scale transcriptome analysis by tiling array and RNA sequencing found loci for more than 10,000 long non-coding RNAs (lncRNAs) in the human genome (1). Although the functions of only a few dozen lncRNAs have been elucidated, several lncRNAs have been found to mediate cell-fate determination, including differentiation, proliferation, senescence, apoptosis, and cancer development (2-5). Moreover, the expression of multiple lncRNAs has been shown to fluctuate according to different biological conditions, such as DNA damage, oncogenic stimuli, and cell differentiation, suggesting the involvement of lncRNAs in biological processes (6-8).
P21-associated ncRNA DNA damage -activated (PANDA) has been identified as an lncRNA induced in response to DNA damage, using super high-resolution tiling array analysis (6). PANDA is located about 5 kb upstream of the transcription start site (TSS) of cyclin-dependent kinase (CDK) inhibitor p21. The genomic DNA of PANDA is composed of one exon and transcribed in the opposite direction of p21, with a 1.5 kb transcription product. The genomic region that includes p21 and PANDA is regulated by tumor suppressor p53 (9), which binds upstream of the p21 TSS and activates both p21 and PANDA transcription in response to DNA damage. PANDA associates with subunit A of nuclear transcription factor Y (NF-Y). NF-Y, a trimeric complex that includes subunits A, B, and C, induces transcription of the apoptosis gene, fas cell surface death receptor (FAS), by binding to its promoter (10). PANDA represses transcription of apoptosis activators, such as apoptotic peptidase activating factor 1 (APAF1), B-cell cll/lymphoma 2-interacting killer (BIK), FAS and LDD, by blocking NF-YA from binding to their promoters, thus repressing apoptosis (6).
The tumor-suppressor protein, p53, controls cell proliferation in response to oncogenic insults and DNA damage (11, 12). Activated p53 triggers cell-cycle arrest, cellular senescence and apoptosis by directly binding to promoters of numerous genes and regulating their transcription, thus preventing DNA-damaged or -stressed cells from proliferating (13). Disruption of the p53 pathway is thought to be a key step in carcinogenesis; and many studies have shown that p53 is frequently mutated in a wide range of human cancer types (14-16). Normally, p53 protein levels are kept low via ubiquitin–proteasome system, which is mediated mainly by murine double minute 2 (MDM2), a ubiquitin ligase (17-19). When cellular DNA is damaged, p53 is phosphorylated through the ataxia–telangiectasia mutated (ATM)/ataxia–telangiectasia and radiation sensitive 3-related (ATR) pathways (20), which disrupts the association of the MDM2-p53 complex, leading to stabilization of p53 protein. p53 is also regulated at translational (21) and transcriptional (22) levels.
p53 has been shown to induce expression of numerous lncRNAs, including lincRNA-regulation of reprogramming (RoR) (23), lincRNA-p21 (7) and PANDA (6). lincRNA-RoR associates with heterogeneous nuclear ribonucleoprotein-I (hnRNP-I) and suppresses p53 translation, which indicates that p53 and lincRNA-RoR form an auto-regulatory feedback loop (23). lincRNA-p21 associates with and recruits hnRNAP-K to target gene promoters and represses transcription, leading to induction of apoptosis (7). Herein, we show that PANDA induced by DNA damage affects stabilization of p53 protein.
Materials and Methods
Cell culture. We used U2OS (human osteosarcoma) cells that were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; GIBCO, Grand Island, NY, USA) in a 37°C incubator with 5% CO2. The cells were exposed to proteosome inhibitor (10 μM) (PEPTIDE INSTITUTE, INC., Osaka, Japan), cycloheximide (100 μg/ml) (SIGMA-ALDRICH, Tokyo, Japan), etoposide (50 μM) (Wako, Osaka, Japan) or vehicle (dimethyl sulfoxide; SIGMA-ALDRICH), as indicated in Figures 2 and 4.
RNA interference (RNAi). U2OS cells were transfected with siRNA oligonucleotides that targeted human PANDA or NF-YA mRNA, using Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer's protocol. Nucleotide sequences of siRNAs for human PANDA were: #1, 5’-AAUGUGUGCACGUAACAGAUU-3’ and #2, 5’-GGGCAUGUUUUCACAGAGGUU-3’. PANDA siRNA#1 was used in Figures 1, 2, 3, and 4. PANDA siRNA#2 was used in Figure 1. Validated siRNA oligonucleotides targeting human NF-YA were obtained from Ambion (siRNA ID: s9528, s9529, s9530; Austin, TX, USA) .
Immunoblotting. Treated cells were lysed with cell lysis buffer (50 mM Tris-HCl pH 7.5, 300 mM NaCl, and 0.3% Triton-X 100) supplemented with protease inhibitors (10 mg/l antipain, 10 mg/l leupeptin, 10 mg/l pepstatin, 10 mg/l trypsin inhibitor, 10 mg/l E64, and 2.5 mg/l chymostatin; PEPTIDE INSTITUTE, INC., Osaka, Japan) and phosphatase inhibitors (1 mM EDTA pH 8.0, 2.5 mM EGTA pH 8.0, 10 mM b-glycerophosphate, 1 mM NaF, and 0.1 mM sodium orthovanadate). The lysates were incubated on ice for 15 min and then centrifuged at 20,600 × g for 15 min at 4°C. The supernatant of each lysate was subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis. The separated proteins were transferred to an Immobilon-PSQ transfer membrane (Merk Millipore, Darmstad, Germany) and subjected to western blot. The following antibodies were used for immunoblotting: anti-p53 (DO-1; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-NF-YA (H-209; Santa Cruz Biotechnology) anti-α-tubulin (SIGMA-ALDRICH). Immune complexes were detected with horseradish peroxidase-conjugated secondary antibodies and an enhanced chemiluminescence system (PerkinElmer, Branchburg, NJ, USA).
Reverse transcription (RT) and quantitative polymerase chain reaction (q-PCR). Total RNA was extracted by an RNeasy Plus kit (Qiagen, Hilden, Germany), according to the manufacturer's protocol. We treated 10 μg of total RNA with Turbo DNase (Ambion); 1.6 μg of DNase-treated total RNA was subjected to RT reactions with oligo(dT)20 primers and SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen); cDNA thus produced was subjected to PCR with Platinum Taq DNA Polymerase (Invitrogen) or q-PCR with QuantiTect SYBR Green PCR Master Mix (Qiagen) and 200 nM gene-specific primers. Primer sequences were: 5’-AGACCCCAGTGGCACCTGAC-3’ and 5’-GGGCAGAACTTGGCATGATG-3’ for PANDA; 5’-CCTCAGCATCTTATCCGAGTGG-3’ and 5’-TGGATGGTGGTACAGTCAGAGC-3’ for p53; and 5’-GCAAATTCCATGGCACCGT-3’ and 5’-TCGCCCCACTTGATTTTGG-3’ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). q-PCR assays were performed using an Mx3000P Real-Time Q-PCR System (Agilent Technologies, Santa Clara, CA, USA) and Rotor-Gene 3000 system (Corbett Research, Sydney, Australia). Relative expression levels of p53 mRNA were normalized to those of GAPDH mRNA.
Results
Several lncRNAs, including X inactive specific transcript (XIST) (24), KCNQ1 overlapping transcript 1 (KCNQ1OT1) (25, 26), antisense IGF2R RNA (AIRN) (27), antisense non-coding RNA in the INK4 locus (ANRIL)/p15 antisense (2, 28-31) and HOXA transcript at the distal tip (HOTTIP) (32), have been shown to affect gene transcription in cis. Therefore, we first examined the involvement of PANDA in regulating p21 expression. We knocked down human PANDA using PANDA-specific siRNA in U2OS cells in which PANDA expression was detectable. RT-PCR assay confirmed that this siRNA effectively reduced PANDA expression (Figure 1A). Immunoblotting assays showed that a substantial decrease in p21 protein was associated with the reduction in PANDA level (Figure 1B). Interestingly, silencing PANDA also reduced the level of p53 protein, which is an activator of p21 transcription (Figure 1B). q-RT-PCR assay showed that p53 mRNA levels were not reduced by silencing PANDA (Figure 1C). These results suggest that PANDA affects p53 protein expression in a trans manner that influences p21 expression.
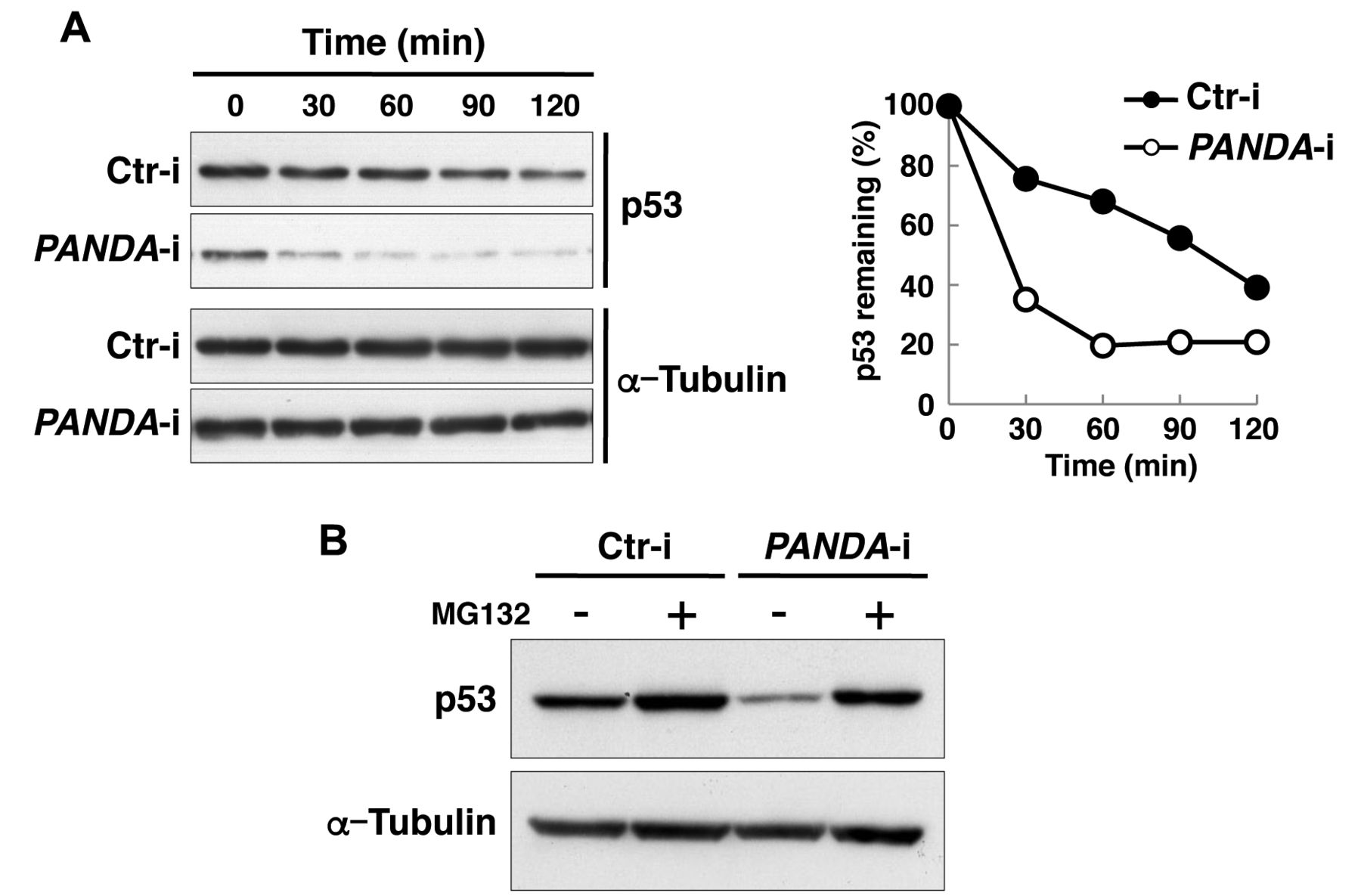
As PANDA depletion reduces p53 protein level, we investigated whether PANDA helps stabilize p53 protein. We measured the half-life of p53 protein in U2OS cells transfected with control and PANDA-targeting siRNA. At 72 h post-transfection with siRNAs, cells were treated with cycloheximide, a protein synthesis inhibitor, for 0, 30, 60, 90 or 120 min and subjected to immunoblot assays. We found p53 protein to be markedly less stable in PANDA-silenced cells (Figure 2A). Treatment with MG132, a proteasome inhibitor, prevented degradation of p53 protein in PANDA-knockout cells (Figure 2B), which indicates that proteolysis of p53 in PANDA-knockout cells is mediated by proteasomes. These results suggest that PANDA contributes to stabilization of p53 protein.

Silencing P21 associated ncRNA DNA damage-activated (PANDA) reduces p53 protein levels. A: U2OS cells were transfected with siRNAs targeting PANDA or control siRNA (Ctr-i) and harvested at 72 h after transfection. The efficiency of PANDA silencing was determined by reverse transcription-polymerase chain reaction (RT-PCR). B: Cells treated as in (A) were subjected to immunoblotting with antibodies against p53, p21 or α-tubulin. C: The effect of PANDA silencing on p53 mRNA levels was determined by quantitative RT-PCR. Data are expressed relative to corresponding values for U2OS cells transfected with control siRNA (Ctr-i). Mean value and standard deviations were calculated from three representative experiments.

Effect of silencing P21 associated ncRNA DNA damage-activated (PANDA) on p53 stability. A: At 72 h post-transfection with siRNAs targeting PANDA or control siRNA (Ctr-i), cells were incubated with cycloheximide (100 μg/ml) for the indicated times. Cell extracts were then immunoblotted with antibodies against p53 or α-tubulin (left panel). Band intensity was measured and normalized to that of α-tubulin. The amount of p53 at 0 min was defined as 100% (right panel). B: At 72 h post-transfection with siRNAs that target PANDA or control siRNA (Ctr-i), U2OS cells were incubated with or without MG132, a proteasome inhibitor (10 μM) for 4 h, after which cell extracts were subjected to immunoblotting as in (A).

P21 associated ncRNA DNA damage-activated (PANDA) regulates p53 protein in an nuclear transcription factor Y subunit alpha (NF-YA)-independent manner. A: U2OS cells were transfected with siRNAs targeting PANDA with or without NF-YA, or with control siRNA (Ctr-i). At 72 h after transfection, cells were harvested and subjected to reverse transcription-polymerase chain reaction to determine the efficiency of PANDA silencing. B: Cells treated as in (A) were immunoblotted with antibodies against p53, NF-YA or α-tubulin.

P21 associated ncRNA DNA damage-activated (PANDA) is involved in the accumulation of p53 protein as a result of DNA damage. A: At 54 h post-transfection by siRNAs targeting PANDA or control siRNA (Ctr-i), U2OS cells were incubated with or without etoposide (50 μM) for 18 h and harvested. Levels of PANDA and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA were determined by reverse transcription-polymerase chain reaction. B: Cell lysates were immunoblotted with antibodies against p53 or α-tubulin.
As PANDA has been shown to repress transcription of apoptosis activators by associating with and inhibiting NF-YA (6), we next examined whether PANDA stabilizes p53 protein through NF-YA. To examine this, we knocked-down both PANDA and NF-YA. RT-PCR and immunoblotting assay confirmed decreased expression of both PANDA and NF-YA (Figure 3A and B). Knocking-down both PANDA and NF-YA also reduced p53 protein to the same level as when knocking-down PANDA alone (Figure 3B), which indicates that PANDA stabilizes p53 protein in an NF-YA-independent manner.
In response to DNA damage, the ATM/ATR pathway phosphorylates p53 protein. This phosphorylation blocks ubiquitination of p53 by MDM2, leading to stabilization of p53 protein that can arrest the cell cycle or induce apoptosis (20). RT-PCR and immunoblotting assay showed that DNA damage induced by treatment with etoposide (topoisomerase II inhibitor) increased the levels of PANDA (Figure 4A) and p53 protein (Figure 4B). Silencing PANDA reduced DNA damage-induced p53 accumulation (Figure 4B). These data suggest that PANDA induced by DNA damage is required in order for p53 protein to accumulate.
Discussion
Several lncRNAs are implicated in regulating transcription, translation and nucleus structure (3, 4, 33). PANDA has been shown to repress transcription of apoptosis activators by inhibiting NF-YA-mediated transcription (6). In this study, we showed a new function of PANDA: stabilization of p53 protein. Reducing PANDA levels resulted in a decrease of p53 protein, but not p53 mRNA level, due to proteolysis of p53 protein by proteasomes. This indicates that PANDA regulates p53 protein post-translationally. p53 degradation in PANDA-knockdown cells was also seen in cells depleted of NF-YA, which suggests that PANDA regulates p53 protein in an NF-YA-independent manner.
The mechanism by which PANDA stabilizes p53 protein is yet to be determined in detail. p53 protein is reportedly ubiquitinated by several ubiquitin ligases, including MDM2 (17-19), p53-induced protein with a RING-H2 domain (PIRH2) (34), ARF-binding protein 1 (ARF-BP1) (35), and constitutive photomorphogenic 1 (COP1) (36), leading to its degradation by proteasome. Because silencing PANDA did not increase expression levels of these ubiquitin ligases (data not shown), PANDA may stabilize p53 protein by repressing the enzymatic activity rather than the expression of these ubiquitin ligases. Phosphorylation of p53 protein by ATM/ATR pathway leads to stabilization of p53 protein by disrupting the association of the MDM2–p53 complex (20). Additionally, the tumor-suppressor protein ARF binds to and antagonizes MDM2 activity, resulting in more stable p53 protein (37-39). The involvement of PANDA in these pathways is an important issue that required further investigation.
We and Hung et al. reported that forced expression of an oncogenic form of small GTPase RAS (called oncogenic RAS) or treatment by doxorubicin (which causes DNA damage) increases PANDA expression (6). In this study, we also showed that treatment with etoposide (which also causes DNA damage) increased PANDA expression. Induction of PANDA by doxorubicin treatment has been shown to require p53 (6). Oncogenic RAS and DNA damage signals may induce PANDA expression through p53.
We demonstrated that silencing PANDA inhibits accumulation of p53 protein induced by DNA damage, which indicates that PANDA is required for p53 to accumulate in response to DNA damage. Our results also imply a regulatory loop between p53 and PANDA, with p53 acting as an upstream inducer of PANDA and PANDA stabilizing p53 protein in positive feedback.
Because the p53 pathway affects cell-fate determination, such as differentiation, apoptosis, senescence and malignant transformation, any lncRNAs that are involved in this pathway, might be pivotal to these processes. Indeed, lincRNA-RoR, which suppresses p53 translation, modulates reprogramming of human-induced pluripotent stem cells (40). Metastasis-associated in lung adenocarcinoma transcript 1 (MALAT1), which represses p53 expression, significantly contributes to malignant lung cancer phenotypes (41-43). lincRNA-p21 induced by p53 affects regulation of the Warburg effects on cancer cells and functions as an oncogene to promote tumor growth (7, 44). PANDA may also contribute to such processes as cancer development, through its place in the p53 pathway. Elucidating the function of PANDA thus warrants further investigation.
Acknowledgements
The Authors thank Chihiro Murasaki and Mika Matsumoto for their technical assistance. We also thank our laboratory members for their helpful discussions. This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan 26430127 (Y. Kotake), 25290044 (M. Kitagawa), 26640073 (M. Kitagawa) and Takeda Science Foundation (Y. Kotake).
- Received November 25, 2015.
- Revision received February 22, 2016.
- Accepted February 24, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Histone H1.2 Represses the Transcription of the p16 Tumor Suppressor Gene
- The lncRNA APOLO interacts with the transcription factor WRKY42 to trigger root hair cell expansion in response to cold
- Stress induced Differential Expression of THAP9 & THAP9-AS1 in the S-phase of cell cycle
- Long Noncoding RNA PANDA Positively Regulates Proliferation of Osteosarcoma Cells