


Abstract
Background: Taxanes and anti-androgen therapies are routinely used for the treatment of metastatic prostate cancer, however the majority of patients eventually develop resistance. Materials and Methods: Eighty kinase inhibitors were screened regarding their ability to inhibit cell viability in CWR22, 22Rv1, PC-3 and DU145 prostate cancer cells using automated toxicity assays. Four kinase inhibitors were selected for further investigation. Results: No significant difference in sensitivity patterns was found between the androgen receptor wild-type CWR22 and its androgen receptor mutant variant 22Rv1, indicating that androgen receptor mutation did not impact on kinase inhibitor sensitivity in this model. Metastatic PC-3 and DU145 prostate cancer cell lines were less sensitive to kinase inhibitors than the non-metastatic CWR22 and 22Rv1. All four cell lines responded to GSK-3 inhibitor BIO, and MEK inhibitor PD198306. DU145 cells were resistant to p75NTR/TrkA and CHK4 inhibitors, RO-082750 and Ryuvidine. Conclusion: Kinase inhibition may be an appropriate strategy for the treatment of prostate cancer.
Prostate cancer is the second most common cancer diagnosed in men globally (1.1 million cases per year), accounting for 15% of all cancer cases in men worldwide (http://globocan.iarc.fr) in 2012. In the United States, the National Cancer Institute (NCI) estimates that 220,800 men will be diagnosed with prostate cancer and 27,540 men will die of cancer of the prostate during 2015 (http://seer.cancer.gov). Several choices exist for the treatment of early prostate cancer, including androgen deprivation therapy, radical prostatectomy, external-beam radiation and prostate brachytherapy, with similar outcomes (1). In patients who develop metastatic disease, androgen deprivation therapy and taxanes remain the main therapeutic strategies. While these treatment approaches extend patient survival, they are not curative and eventually patients develop refractory disease and progress. The taxane family, that includes paclitaxel, docetaxel and the newly-approved cabazitaxel are natural or semi-synthetic plant derivatives widely used in the treatment of castrate-resistant metastatic prostate cancer. Their predominant mechanism of action is through mitotic arrest (2, 3), by inhibiting microtubule dynamics (4). Since 2010 new drugs have been approved for use in patients with metastatic castration-resistant prostate cancer, including androgen receptor inhibitors (abiraterone acetate and enzalutamide), drugs targeting bone metastasis and the microenviroment (alpharadin), immunotherapeutics (Sipuleucel-T) and a new previously-mentioned taxane (cabazitaxel) (5). While these drugs extend life they are not curative and therefore there is a need to explore new potential therapeutic targets for prostate cancer. Kinase inhibitors represent potential agents for the development of a more personalized approach to treating prostate cancer, however clinical trials on inhibitors such as imatinib (PDGF and c-Kit inhibitor) or lapatinib (EGFR and HER2 inhibitor) failed to show benefit in phase II clinical trials, indicating that other kinases may be more appropriate targets (6). Herein, we present preliminary data on the efficacy of 80 kinase inhibitors in vitro against a panel of prostate cancer cell lines. The prostate cancer cell lines tested for cell viability after kinase inhibitor exposure, represent the transition from non-metastatic androgen-dependent (CWR22 - non metastatic, androgen receptor wildtype), to non-metastatic androgen-independent (22Rv1 - non metastatic, androgen receptor) and finally androgen receptor-negative metastatic prostate cancer (PC-3 and DU145 metastatic, androgen receptor negative). This reflects the clinical path where androgen-sensitive localised prostate cancer progresses to castrate-resistant disease, and later to castrate-resistant metastatic disease.
Materials and Methods
Chemicals. The Tocriscreen kinase inhibitor toolbox (Cat. 3514) containing 80 kinase inhibitors was purchased from Bio-Techne (Abingdon, United Kingdom). Unless otherwise stated, all chemicals were obtained from Sigma-Aldrich, Dublin, Ireland.
Cell lines. DU145, PC-3, 22Rv1 and CWR22 were obtained from the American Type Culture Collection (ATCC) (Manassas, VA, USA) and cultured according to recommendations. In brief, CWR22 and 22Rv1 were cultured in RPMI 1640 medium with L-glutamine (Sigma #R8758), and supplemented with 10% fetal bovine serum (FBS) (Sigma #F7524). DU145 was cultured in Minimum Essential Medium (1×) with Earles (Gibco #22561-021, Biosciences, Dublin, Ireland) supplemented with 10% FBS. PC-3 was cultured in F12 Nutrient Mixture (HAM) medium, with L-glutamine (Gibco #21765-029) supplemented with 10% FBS. With the exception of RPMI 1640 and FBS (Sigma), all media and supplements were Gibco (Biosciences, Dublin, Ireland). All media contained 1% of (100X) antibiotic-antimycotic (Life Technologies).
Alamar Blue Assay optimisation and Z-Factor calculation. The seeding density for the four prostate cancer cell lines and Alamar Blue incubation time were optimised to ensure a Z factor of 0.5-1. The Z factor is a statistical method used to determine the robustness of the assay. A Z-factor of 0.5-1 demonstrates a highly robust assay, while a Z-factor of 0-0.5 indicates a below-par robustness (7). CWR22, 22Rv1, PC3 and DU145 were seeded at a range of densities in 96 well plates and treated with 10 μM paclitaxel or 1% DMSO control, followed by an Alamar Blue assay at 72 h post-treatment. Treated cells were incubated with Alamar Blue (93.33 μM) for 2, 4, 6 and 8 h. The Z factor for each treatment was calculated based on values obtained from the entire plate, and values from just the internal wells of the plate. This was to determine if the ‘edge effect’ impacted on the Z-factor. A cell seeding density of 5×103 cells per well, with an incubation time of 8 h with Alamar Blue, and use of the entire plate were selected as the optimal conditions for each cell line.
Prostate cancer cell line toxicity assays. Alamar Blue-based assays were used to assess the effects of kinase inhibitor administration on cell viability. In brief, 1,000 μM of each kinase inhibitor (Panel screen) (Table I) or 0, 150, 250, 500, 1,000 and 1,500 μM (100×) BIO, PD188306, RO-08-2750 or Ryuvidine were pre-loaded into a 96-well plate (Sarstedt, Drinagh, Wexford, Ireland) using the Perkin Elmer Janus Automated Workstation housed in a BigNeat Class II biosafety cabinet. Cells were trypsinized, counted and dispensed by the robotics into a 96-well cell culture plate, at a cell density of 5×103 cells per well. The Janus then performed a series of drug dilutions to yield final appropriate (1×) drug concentrations: 1 in 10 dilution in DMSO, and further diluted 1 in 10 in appropriate media and added to cell culture plates. This was left in culture for 72 h at 37°C in a 5% CO2 incubator. After 72 h Alamar Blue solution was added to the drug-treated plates and incubated for 8 h at 37°C in a 5% CO2 incubator before being read at 530 nm/ 620 nM using a Victor X5 Multilabel plate reader. A percentage viability curve was calculated based on these values. Error was presented at +/− the percentage coefficient variant (%CV). All cytotoxicity assays were conducted in triplicate.
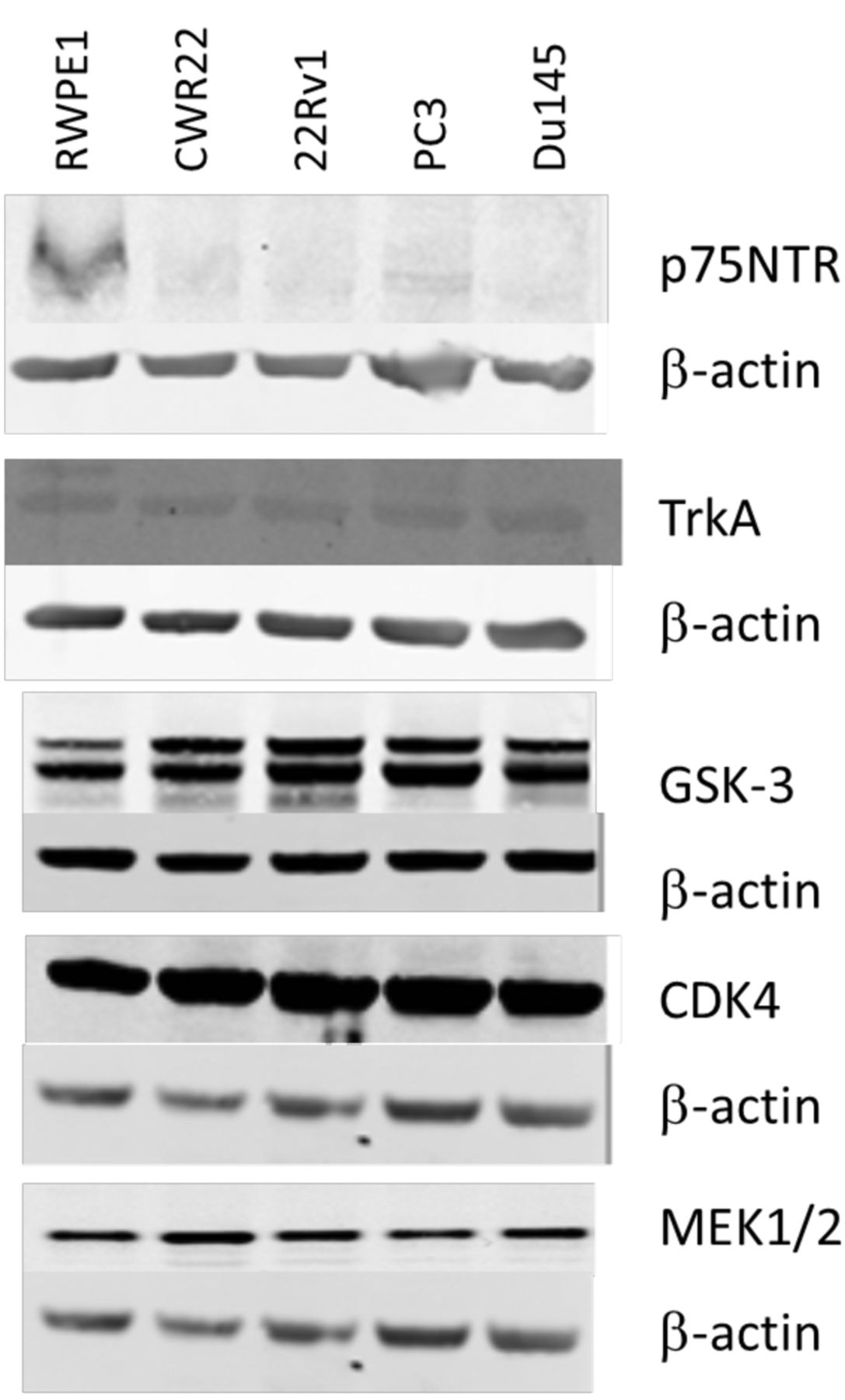
Western blot analysis. Cells were seeded in 10 cm3 dishes at a cell density of 1×106 per dish and allowed to reach 80% confluence. Cells were rinsed twice with cold PBS and lysed directly on the dish with cold RIPA buffer (#89900, Fisher Scientific, Dublin, Ireland) supplemented with protease inhibitors (#78410, Fisher Scientific, Dublin, Ireland), scraped, and spun at 14,000 × g for 15 min at 4°C. Supernatant was collected and stored at −20°C for western blot analysis of protein expression. Extracted proteins were quantified using a BCA kit. The expression of p75NTR, TrkA, GSK-3α/β, CDK4 and MEK1/2 were detected using primary anti-p75NTR rabbit polyclonal antibody #07-476 (Merck Millipore, Cork, Ireland), and anti-TrkA rabbit monoclonal antibody #2508, anti-GSK-3α/β XP rabbit monoclonal antibody #5676, anti-MEK1/2 rabbit polyclonal antibody #9122 and anti-CDK4 mouse monoclonal antibody #2906 (Cell Signalling Technology, Danvers, MA, USA). The anti-p75NTR antibody was diluted 1:1,000, and TrkA antibody 1:1,000, anti-GSK-3α/β antibody 1:1,000, anti-MEK1/2 antibody 1:1,000 and anti-CDK4 antibody 1:2,000 in 5% skimmed milk reconstituted in 1x tris-buffered saline (TBS) (pH8) 0.1% Tween. These dilutions were added to the transfer membrane, and shaken overnight at 4°C, following a 1 h RT blocking in 5% skimmed milk in TBS. Mouse monoclonal anti-β-actin antibody (Fisher Scientific, Dublin, Ireland) (#10624754) was used to confirm equal protein loading. Secondary antibodies used were IRDye 800CW goat anti-rabbit IgG (LI-COR Biosciences, Cambridge, UK) (#926-32211) and IRDye 680LT goat anti-mouse IgG (LI-COR Biosciences) (#926-68020) and detection was imaged on the LI-COR ODYSSEY CLx imaging system.
Statistical analysis. Data analysis was performed using the GraphPad Prism Version 5. All statistical tests were two-sided, and an association was considered statistically significant when p-values were less than 0.05. For the kinase panel screen a two-way ANOVA with Bonferroni's multiple comparisons test was used to determine whether there were significant differences in the sensitivity of different cell lines to a particular compound.
Results
Comparison of effect of androgen independence on sensitivity to kinase inhibition. We performed a cell viability screening for 80 kinase inhibitors in CWR22 and 22Rv1 prostate cancer cells measuring the mean % cell viability compared to control after 72 h exposure to the drugs. CWR22 is the parent cell line and is androgen receptor-positive and androgen responsive. The 22Rv1 was generated from a castrate-resistant xenograft model of CWR22 (8) and has a mutated androgen receptor and is androgen independent (9). Figure 1A and B show the sensitivity profiles of CWR22 and 22Rv1 to the 80 kinase inhibitors. Performing a two-way ANOVA with a Bonferroni post-hoc test to compare the mean inhibition of cell proliferation by each compound between the two cell lines, no significant difference was found between the CWR22 and 22Rv1 for any of the compounds tested. This suggests that the acquisition of androgen independence in this model has not altered the sensitivity of the CWR22 cell line to kinase inhibition and suggests that kinase inhibition may be a useful strategy for the treatment of prostate cancer before or after castrate resistance emergence, in the context of androgen receptor mutation.
Kinase inhibitors used in screening and their main kinase target.
Metastatic PC-3 and DU145 prostate cancer cell lines showed a decreased range of sensitivity to kinase inhibition compared to non-metastatic CWR22 and 22Rv1. The metastatic PC-3 and DU145 prostate cancer cell lines were derived from a bone metastasis and a brain metastasis respectively (10, 11). Both cell lines are androgen receptor-negative and are commonly used as in vitro models of castrate-resistant metastatic prostate cancer. Figure 1C and D show the sensitivity profiles of PC-3 and DU145 to the 80 kinase inhibitors. Performing a two-way ANOVA with a Bonferroni post-hoc test to compare the mean inhibition of cell proliferation by each compound between the CWR22 and PC-3 cell lines, the PC-3 cell line showed statistically significant resistance to 14 compounds that targeted a range of kinases including AMPK, Chk1, GSK-3, IGF1R, JAK3, MEK, PKB, PKC, Src and VEGFR (Table II). Comparing CWR22 and DU145 cell lines, the DU145 cell line showed statistically significant resistance to 17 compounds that targeted kinases including BTK, Chk1, EGFR, GSK-3, IGF1R, JAK3, JNK, p38MAPK, MEK, PKA, PKB, PKC, and TrkA (Table III). Table IV shows kinase inhibitors that exhibit a significant difference in response between the PC-3 and DU145 cell lines.

% Cell Viability after 72 h exposure to Tocriscreen kinase inhibitors in (1A) CWR22, (1B) 22Rv1, (1C) PC-3 and (1D) DU145 prostate cancer cell lines.
Kinase inhibitors displaying a significant difference in their ability to inhibit cell viability in AR positive non-metastatic CWR22 versus AR-negative bone metastatic PC-3.
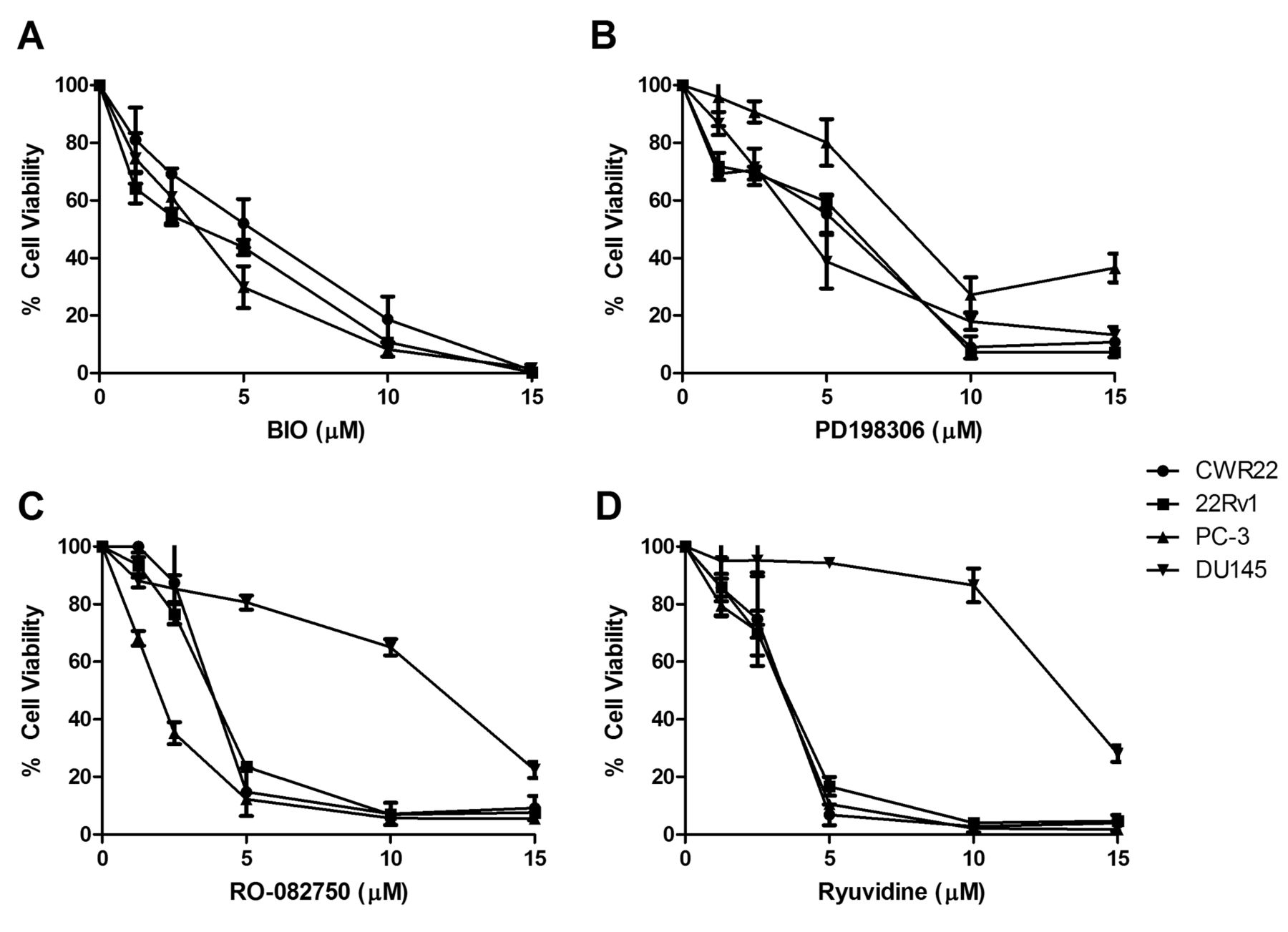
Confirmation of expression of kinase inhibitor targets TrkA, p75NTR, GSK3α/β, MEK1/2 and CDK4 in prostate cancer cell lines. To validate the findings of the 80-panel kinase screen, four kinase inhibitors were chosen for further exploration. Three inhibitors had shown activity against all cell lines tested and include BIO which targets GSK-3, PD198306 which targets MEK, and Ryuvidine which targets CDK4 (Figure 1). Additionally we also chose RO-082750 which targets p75NTR and TrkA, and has shown sensitivity against CWR22, 22Rv1 and PC-3, but limited response in DU145 in our screening (Figure 1). Western blotting was performed to confirm the expression of p75NTR, TrkA, GSK-3, MEK1/2 and CDK4 in the prostate cancer cell lines, as shown in Figure 2. To determine the effects of increasing doses of the drugs on prostate cancer cell line viability we performed toxicity assays for the 4 compounds on each cell line. All four cell lines showed a similar response to the GSK-3 inhibitor BIO (Figure 3A), and the MEK inhibitor PD198306 (Figure 3B). CWR22, 22RV1 and PC-3 were also sensitive to the p75NTR and TrkA inhibitor RO-082750 (Figure 3C), and the CHK4 inhibitor Ryuvidine (Figure 3D), while DU145 was resistant to both.
Kinase inhibitors displaying a significant difference in their ability to inhibit cell viability in AR positive non-metastatic CWR22 versus AR-negative brain metastatic DU145.
Kinase inhibitors that display a significant difference in their ability to inhibit cell viability in bone metastatic PC-3 versus brain metastatic DU145.
Discussion
We performed screening of 80 kinase inhibitors against a panel of prostate cancer cell lines, in order to identify potential kinase inhibitor targets that may have therapeutic potential in prostate cancer. We found that prostate cancer cell lines were sensitive to a large range of kinase inhibitors, with the CWR22 and 22Rv1 cell lines showing the sensitivity to a larger number of kinase inhibitors than the metastatic prostate cancer cell lines PC-3 and DU145. We have previously reported that the DU145 and PC-3 cell lines, are more resistant to docetaxel and our microtubule inhibitor EL102 compared to CWR22 and 22Rv1(12), indicating that the PC-3 and DU145 have intrinsic resistance to certain drugs.
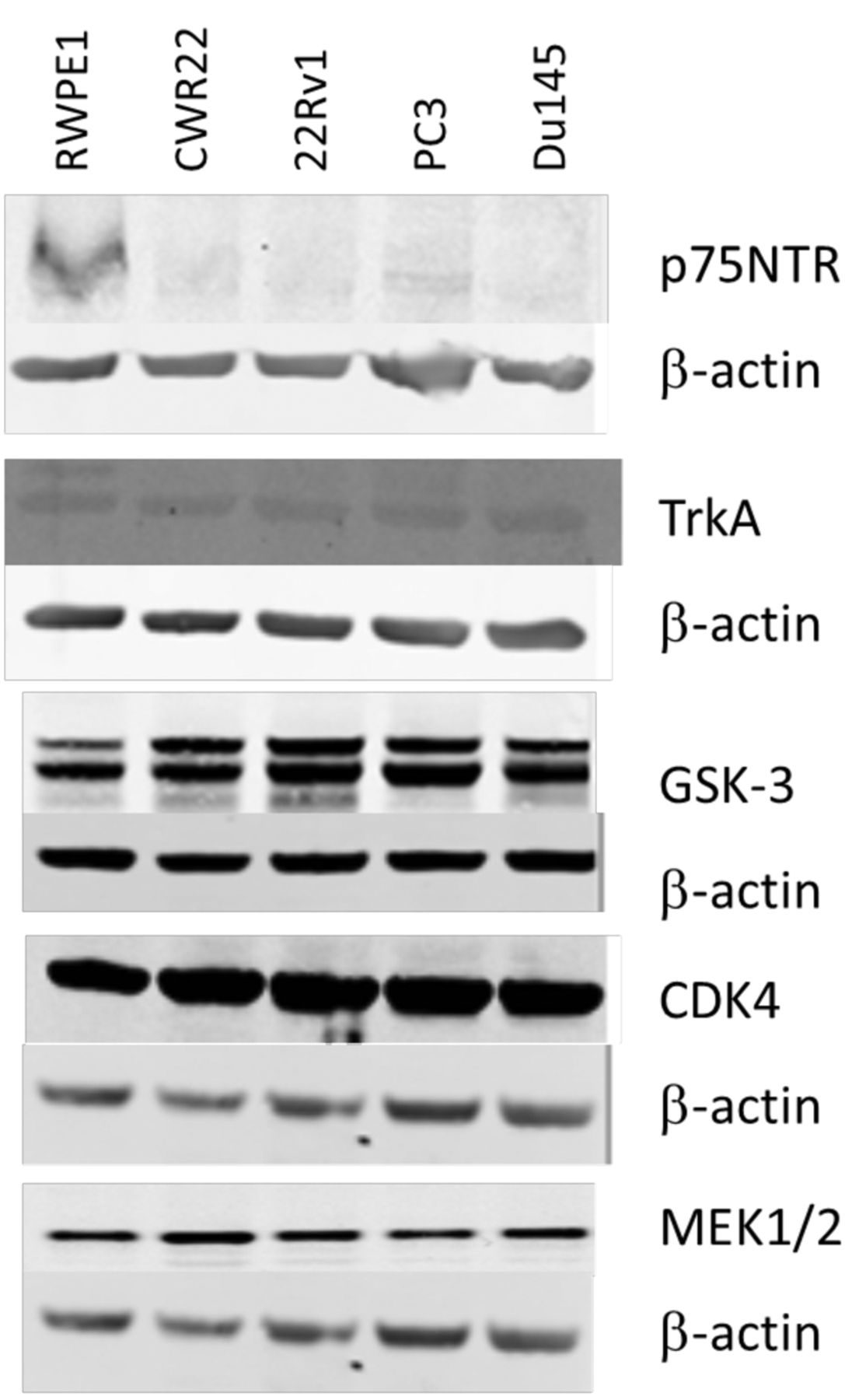
Expression of p75NTR, TrkA, GSK-3, CDK4 and MEK1/2 in CWR22, 22Rv1, PC-3 and DU145 prostate cancer cell lines.
One of the main obstacles to the treatment of advanced-stage prostate cancer is the emergence of castrate-resistant prostate cancer. Castrate resistance can emerge through several mechanisms, one of which is through mutation of the androgen receptor. This can include mutations in its ligand binding domain that alter its structure allowing the androgen receptor to be activated by alternative ligands (13). Splice variants can also emerge that lack the ligand binding domain resulting in constitutive activation of the androgen receptor (13). The 22Rv1 cell line contains two androgen receptor isoforms not present in the parent CWR22, the larger of which contains a tandem duplication of exon 3, resulting in the addition of 39 amino acids in the DNA-binding domain, resulting in increased receptor promiscuity (14). A second smaller isoform represents a carboxy-terminally truncated variant that lacks the ligand binding domain (14). Our results show that the acquisition of these mutations in 22Rv1 did not alter the sensitivity to the kinase inhibitors tested compared to CWR22. This is highly relevant, as it suggests that kinase-targeted therapeutics may be equally effective in pre- and post-castration resistance, when mediated by these types of androgen mutation.
While the androgen receptor-negative metastatic PC-3 and DU145 prostate cancer cells do respond to several of the kinase inhibitors tested, they were found to be more resistant to some AMPK, Chk1, GSK-3, IGF1R, JAK3, MEK, PKB, PKC, Src and VEGFR inhibitors (PC-3) and including BTK, Chk1, EGFR, GSK-3, IGF1R, JAK3, JNK, p38MAPK, MEK, PKA, PKB, PKC, and TrkA inhibitors (DU145), when compared to CWR22. Whether this is due to a lack of expression of the androgen receptor, the development of metastatic potential or to different genetic backgrounds cannot be answered within the limits of this study. Both PC-3 and DU145 were resistant to the JAK3 inhibitors ZM449829 and ZM39923 that have cross-reactivity with JAK1, EGFR and CDK4. Both JAK1 and JAK3 activate STAT3 signaling. While the role of JAK3 is not currently known in prostate cancer, the DU145 cell line has previously be found to be resistant to JAK1 inhibition (15). Ueda et al. previously showed that JAK inhibition in androgen receptor-positive LNCaP prostate cancer cells can result in inhibition of androgen receptor activation via STAT3 inhibition, which may explain in part why the CWR22 and 22Rv1 are more responsive to JAK3 inhibition (16).
We chose to further explore the expression and dose-response profiles of the targets of three inhibitors which had shown activity against all cell lines tested including BIO which targets GSK-3, PD198306 which targets MEK, and Ryuvidine which targets CDK4. Additionally we also chose RO-082750 which targets p75NTR and TrkA, and had shown sensitivity against CWR22, 22Rv1 and PC-3, but limited response in DU145 in our screening. All prostate cancer cell lines expressed GSK-3, that has recently been proposed as a potential chemoprevention target for prostate cancer (17). GSK3α is mainly expressed in low-risk prostate cancer, while GSK3β is mainly expressed in high-risk prostate cancers. Cytoplasmic levels of GSK-3β correlated with increased clinical stage, lymph node metastasis, extracapsular extension, and high Gleason score, all features of an aggressive clinical course (18). Gao et al. explored the functional effects of GSK3α and GSK3β in prostate cancer. GSK3α promotes proliferation and resistance to apoptosis, while GSK3β promotes cell migration, and inhibits cell-cell contacts via activation of epithelial to mesenchymal transition (19). Additionally GSK3β inhibition has been shown to deplete prostate cancer stem cells (20). All cell lines also expressed MEK1/2. The Raf/MEK/ERK signaling cascade is a key player in prostate cancer progression, and is activated via Akt during the transition to androgen independence (21). Increased expression of ERK which is activated by MEK is associated with increased risk of biochemical recurrence, indicating that inhibition of this signaling pathway may be a good target for the prevention of prostate cancer progression (22). The third inhibitor explored targeted CDK4. CDK4 inhibition in prostate cancer cells results in G0/G1 arrest (23). Artemisinin which has been shown to have anti-proliferative activity against prostate cancer, was found to exert its effects via inhibition of CDK4 (24). Interestingly CDK4/6 inhibitors have been shown to reverse the resistance conferred by the F876L androgen receptor mutation in prostate cancer against enzalutamide (25). Additionally we explored the TrkA and p75NTR inhibitor RO-082750. In prostate cancer p75NTR is predominantly expressed by the stromal tissue and was decreased in the periphery basal cells of tumor foci (26). Similarly we observed a down-regulation of p75NTR in our prostate cancer cell lines compared to the normal immortalized RWPE1. TrkA was expressed in all prostate cancer cell lines tested at similar levels. Previous research by Festuccia et al. suggests that TrkA may play a role in prostate cancer progression, as blockade with the TrkA inhibitor CEP-701 inhibited cell migration and invasion in prostate cancer cells (27). DU145 have previously been shown to express large quantities of NGF the ligand for TrkA and P75NTR (28), which may contribute to the increased resistance that we observed.
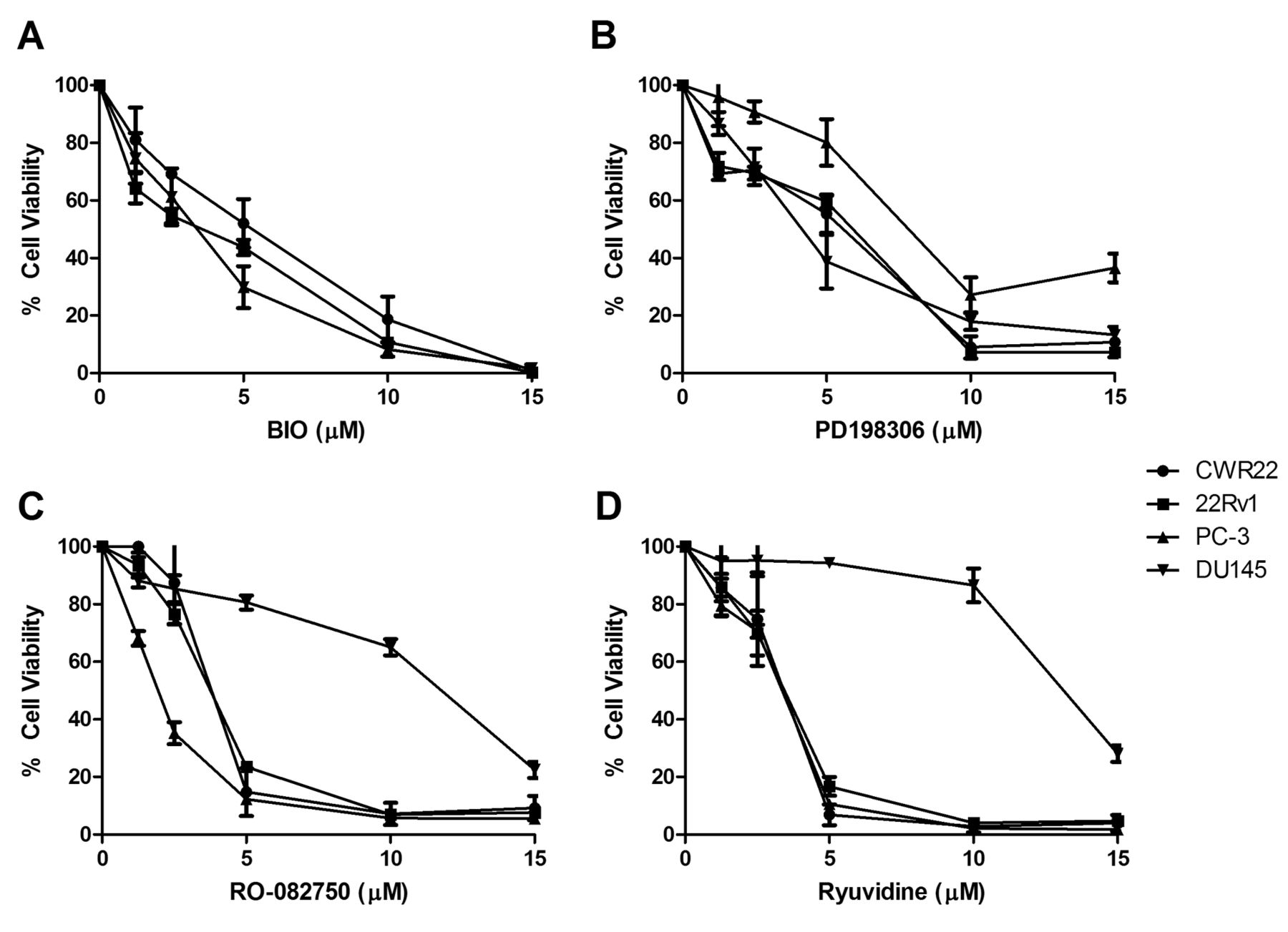
Dose-response effects of the (3A) GSK-3 inhibitor BIO, (3B) MEK1/2 inhibitor PD198306, (3C) TrkA & p75NTR inhibitor RO-082750 and the (3D) CDK4 inhibitor Ryuvidine in CWR22, 22Rv1, PC-3 and DU145 prostate cancer cell lines.
There is no current cure for castrate-resistant metastatic prostate cancer. Novel adjuvant chemotherapies are continually being developed to address this fact, with the approval of six new agents since 2010. The emergence of resistance remains a problem which can be a result of acquired alterations in androgen receptor signaling or other mechanisms including multiple drug resistance protein pumps such as MDR1, BCRP, MRP1, MDR2, altered growth factor receptor pathway activation (e.g. IGFR, VEGFR, EGFR), hypoxia-related resistance, tubulin mutation and altered tubulin isoform expression, and NFκB activation (29, 30, 31). For this reason it is important to explore alternative therapeutic targets that can be used either alone or in combination with current therapeutics to improve patient outcomes. The results of the present study demonstrate that prostate cancer cells are sensitive to a range of kinase inhibitors. While the range of inhibitors that displayed activity were greater in the androgen receptor-positive CWR22 and 22Rv1s, than in the androgen receptor-negative PC-3 and DU145, several kinase inhibitors displayed activity in all cells lines. This suggests that at least in cell line models of cancer progression that there are kinases that are important drivers of prostate cancer proliferation independent of androgen receptor status. These kinase inhibitors could have efficacy as therapeutic agents across all stages of prostate cancer progression, allowing us to target proliferation at both the early stage and later upon metastatic progression. Another approach is to develop kinase inhibitors that are particularly sensitive against castrate-resistant bone metastatic prostate cancer, that is represented by the PC-3 cell line. Figure 1 demonstrated PC-3 sensitivity to Aminopurvalanol A (CDK), BIO (GSK3α/β), ER 27319 maleate (Syk), Ryuvidine (CDK), IMD 0354 (IKK), Ro 082750 (TrkA), BI 78D3 (JNK), IKK 16 (IKK), PHA 665752 (cMET), which we confirmed for Ro 082750 and BIO. Syk inhibitors in particular would be of interest as Syk SNPs are associated with prostate cancer risk (32), and exhibits increased expression with progression, with Syk siRNA knockdown resulting in inhibition of invasion and bone metastasis in murine models of prostate cancer (33). IMD 0354 and IKK 16 both target IκB kinase-β (IKKβ), resulting in the inhibition of NF-κB translocation to the nucleus. NF-κB gene signatures are predictive of prostate cancer progression (34), and metastasis (35), indicating that targeting this pathway may be beneficial for the prevention of metastatic progression. Future studies should examine their potential to enhance the effects of the existing therapeutics and determine their potential as single agents or as combination therapies.
Acknowledgements
This work was supported by Galway University Foundation (RNR1008), Irish Cancer Society (PCT13MCD), Breast Cancer Now (2013MayPR019), Cancer Care West-Hardiman Scholarship, and the NCBES Screening Core by PRTLI5 Advancing Medicine through Discovery programme.
- Received November 11, 2015.
- Revision received December 15, 2015.
- Accepted January 4, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.