


Abstract
Background: Ovarian cancer is difficult to treat due to absence of selective drugs and tendency of platinum drugs to promote resistance. Combination therapy using epigenetic drugs is predicted to be a beneficial alternative. Materials and Methods: This study investigated the effects of combination therapies using two structurally different histone deacetylase (HDAC) inhibitors (HDACi), sodium butyrate and suberanilohydroxamic acid (SAHA), with the calpain inhibitor calpeptin on two characteristically different ovarian cancer cell lines, CAOV-3 and SKOV-3. Results: Suboptimal doses of HDACi and calpeptin produced several effects. Growth inhibition was enhanced and the epigenetically silenced tumor suppressor genes ARHI, p21 and RARβ2 were re-expressed. Methylation of specific CpG residues in ARHI were reduced. Cell-cycle progression was inhibited and apoptosis, as well as autophagy, were induced. The phosphorylation of ERK and Akt were differentially effected by these inhibitors. Conclusion: The re-expression of tumor suppressors may sensitize ovarian cancer cells, which then undergo apoptosis and autophagy for cell death.
- Ovarian cancer
- combination therapy
- epigenetic drugs
- histone deacetylase inhibitors
- calpain
- methylation
- cell cycle
- apoptosis
- autophagy
Ovarian cancer accounts for the greatest number of deaths out of all female reproductive system malignancies (1). In 2016, The American Cancer Society estimates 22,280 ovarian cancer diagnoses and 14,240 ovarian cancer deaths in the United States (2). The risk of a woman developing ovarian cancer in her lifetime is 1-2 out of 100. Since tumorigenesis portrays an asymptomatic manifestation, only 25% of ovarian cancers are found in early stages. Clinically, the only serum biomarker molecule used to for monitoring progression and regression is cancer antigen (CA)-125, which is elevated in about 80% of women with late stage ovarian cancer. However, this has no adequate specificity or sensitivity for early detection (3).
Ovarian carcinomas consist of heterogeneous subtypes, which commonly include serous, endometrioid, clear cell and mucinous cancerous subtypes. These are further classified into benign, intermediate and malignant clinical manifestations. The diverse characteristics of ovarian carcinoma arise through inheritance or sporadic mechanisms, although the latter are still not fully understood. Shared genetic markers seen in cancers, such as breast cancer (BRCA)1 and 2, estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor (HER)2 and aplysia Ras homology member 1 (ARHI) in breast cancers, are also involved in ovarian cancers. Additionally, miRNA regulation and epigenetic gene silencing are altered in ovarian cancer in ways similar to those observed in breast cancers (4). All together, these lines of evidence indicate a similar origin of both types cancers.
Nevertheless, current standard chemotherapies are administered based on the stage and grade of the cancer, rather than the histotype. For example, platinum-based therapy has been the mainstay for the past four decades, regardless of histologic subtypes, mutational status or biomarkers (5). Its cytotoxic effect involves the active cellular uptake of cisplatin, which binds to DNA and produces intra- and inter-strand crosslinks that break the DNA. This results in apoptosis and cell death (6). However, a major challenge with present therapies is chemoresistance. Resistance to platinum therapy is associated with the limited formation of platinum-DNA arrangements and the prevention of cell death after the platinum-adduct formations. Relapse within 6 months of completing the first-line therapy classifies a patient as “platinum-resistant” and this develops in 30-40% of women (7). Therefore, novel therapeutic targets and treatments are necessary to successfully treat patients.
Although cancer therapeutics primarily target genetic alterations, in recent years, epigenetics has been proposed as a new target to overcoming drug resistance (8). In particular, epigenetic alterations in genetically predisposed cells may initiate the development of these cells into cancer progenitors, which are often resistant to traditional therapies (5, 9). This mechanism could explain why two people with identical genetic signatures, such as the BRAC1 mutation, have different rates of cancer progression. Additionally, this suggests that a combination therapy, which includes epigenetic drugs, should reduce cancer relapse and be effective on drug resistant cancer cells (10-12). Furthermore, since tumor cells reversibly transform via the epithelial mesenchymal transition (EMT) and mesenchymal epithelial transition (MET), which is necessary for metastasis and may involve epigenetic mechanisms, combination treatment may also be useful against metastasis (5, 13). Metastasis is a step-wise reversible process that increases the expression of cell surface receptors that facilitate motility and reduces the expression of proteins that promote adhesion (EMT). After becoming mobile, cells must reverse this phenomenon (MET) to anchor in a new tissue and reversible epigenetic processes may regulate this transition (13). Thus, the reversal of epigenetic alterations in cancer progenitor cells should sensitize these cells, as well as facilitate growth inhibition, block metastasis and reduce the likelihood of relapse.
Accordingly, DNA methyltransferase 1 (DNMT1) inhibitors and histone deacetylase inhibitors (HDACi) can re-sensitize resistant ovarian cancer cells to cisplatin via the demethylation and re-expression of RGS10, a regulator of cell survival and chemoresistance (14). Additionally, HDACi promote the degradation of DNMT1, which results in CpG demethylation and the re-expression of tumor suppressor genes (TSG) p21, p16 and retinoic acid receptor beta 2 (RARβ2) (15). Re-expression of TSG by HDACi possibly sensitizes cancers cells to lower doses of other cytotoxic agents (10). Indeed, the combination of HDACi with TRAIL inhibitory antibodies reduced breast tumor size in a xenograft mouse model (16). We have previously shown that the combination of HDACi with telomere inhibitor GT-oligo produced enhanced growth inhibition in different types of ovarian cancer cells (17). In addition, the combination of HDACi with calpain protease inhibitor produced more than additive effects in inhibiting several types of cancer cells including, breast cancer, ovarian cancer, prostate cancer and leukemia (3, 19). Calpain is a ubiquitous protease, which regulates many types of signaling proteins and pathways. Alteration in calpain expression and activity has been observed in many diseases; calpain inhibition has been implicated in the therapy of cardiovascular diseases (20) and cancer (3, 19, 21).
In this study, we investigated the effects of two structurally different HDACi (Class I sodium butyrate (SB) and Class II suberanilohydroxamic acid (SAHA)) in combination with the calpain protease inhibitor calpeptin on ovarian cancer cells. Two discrete ovarian cancer cell lines (CAOV-3 and SKOV-3) were examined. We found that the combination therapy produced enhanced growth inhibition in these two discrete ovarian cancer cell lines. Additionally, we identified the inhibitors' mechanisms of actions. The inhibitors (i) inhibit cell cycle progression, (ii) decrease metastatic activity, (iii) induce apoptosis, (iv) promote autophagy and (v) result in the re-expression of TSG, including ARHI, p21 and RARβ2. We also identified differential demethylation of ARHI in both cell lines. Finally, (vi) we observed differential inhibition of two important signaling pathways – v-ask murine thyomoma viral oncogenes homolog 1 (Akt) and extracellular signal-regulated kinase (ERK). These findings reveal the mechanisms by which a combination therapy effectively treats ovarian cancer cells and validate the usefulness of this combination therapy for clinical purposes.
Materials and Methods
Reagents. SB and SAHA were purchased from Sigma (St. Louis, MO, USA) and calpeptin from Nova Chemicals (Moon Township, PA, USA). RPMI-1640 and Dulbecco's modified Eagle's medium were obtained from Invitrogen (Carlsbad, CA, USA). Protease inhibitor cocktail tablet was from Roche, Mannheim, Germany. The Annexin/PI apoptosis analysis kit was from BDR (San Diego, CA, USA). Genomic DNA was isolated using a kit obtained from Stratagene (LaJolla, CA, USA). Bisulphite reactions kit and the cDNA preparation kit were obtained from Qiagen (Valencia, CA, USA). BD Matrigel invasion chambers were from BD Biosciences (Bedford, MA, USA). The SYBR green RT-PCR mix was purchased from Applied Biosciences (Foster City, CA, USA).
Cell culture. CAOV-3 and SKOV-3 ovarian cancer cells were obtained from ATCC, Manassas, VA, USA. Cells were cultured in RPMI-1640 media, containing 100 IU/ml penicillin, 100 μg/ml streptomycin and 50 ml 10% heat-inactivated fetal bovine serum. At 70-80% confluence, cells were exposed to dimethyl sulfoxide (control) or distinct concentrations of drugs (SB, SAHA and/or calpeptin). Cells were incubated for different times before being harvested.
Cell survival assay. Viable cells were counted using a trypan blue exclusion assay in six-well plates. The cells were trypsinized and 20-μl cell suspensions were mixed with 20 μl trypan blue. The unstained cells (viable) were counted and the percentage was plotted against time.
List of primers used for RT-PCR.
In vitro cell migration assay. A chemotaxis assay was used to test the effects of inhibitors on cell migration. The assay was performed in 24-well plates and contained transwell chambers with 8 μM pore membranes. Cells were treated with specific concentrations of inhibitors, harvested two days later by trypsinization and washed in serum-free RPMI growth medium. The upper chamber received 200,000 cells suspended in 500 μl serum-free RPMI growth medium and the lower chamber received 0.5 ml of conditioned media from the actively growing untreated ovarian cancer cells. The conditioned media was used as a chemoattractant. After 12 h, cells from the chambers were aspirated and wiped with cotton swabs. The membranes were stained with Diff-Quik Stain (IMEB Inc., San Marcos, CA, USA) and the migrated cells were viewed under a light microscope and counted. Results were presented from triplicate sets as percent motility of the control untreated cells as 100%.
Methylation specific sequence analysis. Ovarian cancer cells were cultured in the presence and absence of inhibitors (1 mM SB and 10 μg/ml calpeptin, respectively) for 48 h. Following this, genomic DNA was isolated from cells and bisulfite treated. Purified DNA was sent to EpigenDX, Worcester, MA, USA, for methylation specific sequencing of the ARHI gene. Assays were standardized using positive and negative controls; the percent of methylation at specific CpG sites was determined.
Quantitative polymerase chain reaction (qPCR). Ovarian cancer cells were grown in 10-cm plates, treated with inhibitors and harvested. Total RNA was isolated using Trizol reagent (Invitrogen), according to the manufacturer's protocol. RNA was converted to cDNA after digesting genomic DNA contamination. Real-time quantitative PCR was performed using prepared cDNA (Table I). Results were normalized to the expression level of β-actin (ACTB) transcripts within each sample with the same primers. PCRs were performed in triplicates, and results are presented as the mean and standard deviations.
Cell cycle and apoptosis assays. Ovarian cancer cells were treated with inhibitors. Untreated control cells and treated cells were washed with phosphate buffered saline (PBS). For cell cycle analysis, cells were fixed with a medium containing 35% ethanol for 5 min at room temperature. The cells were stained for 30 min in the dark with 25 μg/ml propidium iodide (PI), containing 50 μg/ml RNAse in PBS, before FACS analysis. The apoptosis assay was performed with the annexin/PI assay. After treatment, untreated and treated cells were harvested and suspended in 100 μl 1x binding buffer. Then, 5 μl annexin reagent conjugated to fluorescein isothiocynate (FITC) and 5 μl PI were added to each sample as described in the kit protocol (BD Biosciences). Samples were kept in the dark for 30 min at room temperature. Finally, 900 μl 1x binding buffer was added to each sample and the dual fluorescence of FITC and PI were measured during FACS analysis.
Immunostaining procedure and autophagy assay. Ovarian cancer cells were grown in 8-well plates and treated with inhibitors. Media was aspirated from each well and the cells were fixed for 15 minutes with 200 μl 4% paraformaldehyde at room temperature and washed with PBS 3 times. Next, cells were treated for 30 min with 200 μl 0.1% Triton X100 and 0.1% sodium citrate at room temperature. The solution was aspirated and the wells were washed 3 times with PBS and blocked for 1 hour with 300 μl bovine serum albumin (BSA) at room temperature. The solution was aspirated and the wells washed 3 times with PBS. The primary anti-LC3b was added in 3% BSA to the wells and incubated at 4°C overnight. The solution was then aspirated and the wells were washed 3 times with PBS. An appropriate dilution of secondary antibody in 3% BSA (conjugated to FITC) was added and incubated for one hour at room temperature. The solution was aspirated and the wells washed 3 times with PBS. Then, 100 μl DAPI (Invitrogen) was added to each well and incubated for 5-10 min. The solution was aspirated and the wells washed 3 times with PBS. A drop of mounting solution (Prolong Gold; Invitrogen) was added to each slide and a cover slip placed to hold in position. Each slide was sealed with nail oil, dried, viewed and photographed under fluorescent microscope.
Protein immunoblot analysis. Ovarian cancer cells were grown in 10-cm plates and treated with inhibitors. The cells were harvested with trypsin/EDTA, washed with PBS and collected by centrifugation at 1,000 rpm. Next, cells were lysed with ice cold 1% NP-40 lysis buffer, including vanadate and protease inhibitor cocktail tablet (1 tablet/10 ml buffer). For each sample, protein concentrations were calculated with BSA as the control, using a protein assay reagent (Bio-RAD, Hercules, CA, USA). Equal amounts of protein from each sample were resolved by SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked with 3% non-fat milk powder or 5% BSA for phospho blots in Tris-Buffered Saline/Tween (TBST) and then incubated with the primary antibody at 4°C overnight. Each membrane was washed with TBST 3 times for 10 min. The secondary antibody conjugated to horseradish peroxide, was added and the membrane was incubated for 1 hour at room temperature. The membrane was washed with TBST 3 times for 10 min, developed using a chemiluminescent reagent and photographed using Image Quant (GE LAS4000; Piscataway, NJ, USA).

Growth inhibition and morphological changes of CAOV-3 and SKOV-3 ovarian cancer cells by HDACi and calpeptin. A. CAOV-3 ovarian cancer cells were treated with 0.5 mM sodium butyrate (SB), 2 μM suberanilohydroxamic acid (SAHA), 5 μg/ml calpeptin (Calp) or combination of SB and Calp (p-value=0.0002) and SAHA and Calp (p-value=0.0001), at the same concentrations. B. SKOV-3 ovarian cancer cells were treated with 1 mM SB, 10 μM SAHA, 10 μg/ml Calp or combination of SB and Calp (p-value=0.0001) and SAHA and Calp (p-value=0.0001), at the same concentrations. Viable cells were washed, trypsinized and counted after four days. All treated cells are expressed as a percent relative to the arbitrarily set 100% of the dark shaded column of control cells. C. CAOV-3 ovarian cancer cells were treated with 1 mM SB, 10 μM SAHA, 20 μg/ml Calp or a combination of SB and Calp, as well as SAHA and Calp, at the same concentrations. D. SKOV-3 ovarian cancer cells were treated with 1 mM SB, 10 μM SAHA, 20 μg/ml Calp or a combination of SB and Calp and, also, SAHA and Calp, at the same concentrations. Morphology changes and number of cells are different in the cells with inhibitors when compared to control cells. Photographs were taken after four days of treatment.
Results
Combination treatment effects on growth inhibition and morphology. To test the efficacy of the combination therapy, two characteristically different ovarian cancer cells lines, SKOV-3 and CAOV-3, were employed in this study. Cells were treated with sub-optimal levels of chemically distinct HDACi (SB and SAHA) and calpain protease inhibitor, calpeptin (Calp). The combination treatment of HDACi and calpeptin resulted in statistically significant 80-90% growth inhibition after 4 days (Figure 1A and B; SB/Calp p<0.0002; SAHA/Calp p<0.0001 for CAOV-3 and p<0.0001 for SKOV-3). The effective dose of each inhibitor was lower in combination than the effective doses of the inhibitors when used as a single agent (not shown).
Distinct morphological changes were observed both in CAOV-3 and SKOV-3 ovarian cancer cells after treatment with the inhibitors. After 4 days, cells decreased in number and assumed a more rounded shape with the addition of HDACi and calpeptin, which suggests cell death (Figure 1C and D). The distortion of cell structure in both ovarian cancer cell lines was more evident when treated with calpeptin.
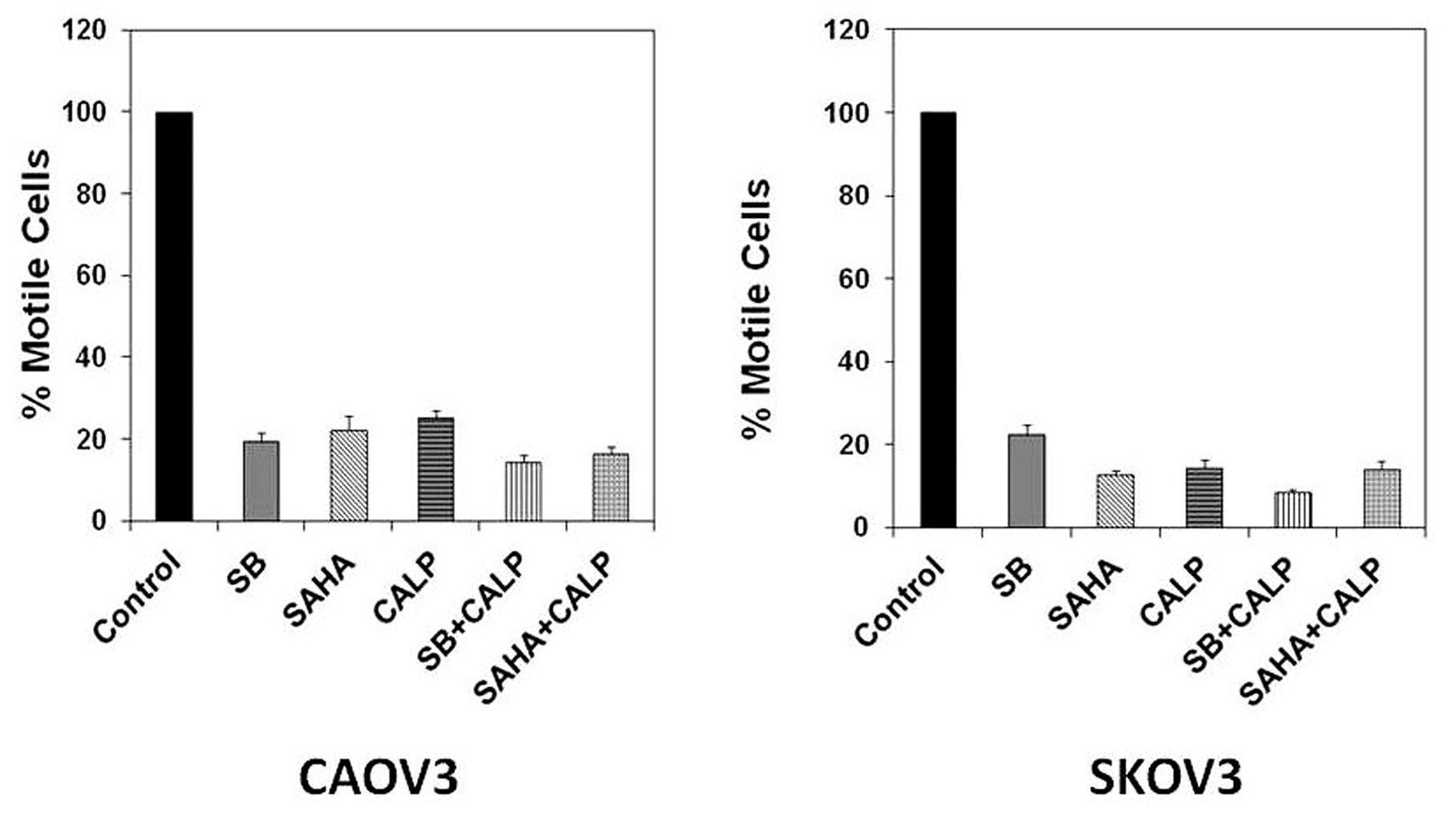
Inhibition of motility by HDACi and calpeptin. The effects of HDACi and calpeptin on the invasive nature of ovarian cancer cells was also examined. As a marker of metastatic potential, the degree of motility of ovarian cancer cells was assessed by an in vitro cell migration (transwell) assay (13). The number of untreated cells that moved across the membrane was assigned as control cells, being 100% motile. The percent of treated cells were plotted and approximately 70-90% reduction in motility of both CAOV-3 and SKOV-3 ovarian cancer cells was observed (Figure 2A and B).

Transwell inhibition of CAOV-3 and SKOV-3 ovarian cancer cell motility. A. CAOV-3 ovarian cancer cells were treated with 0.5 mM sodium butyrate (SB), 2 μM suberanilohydroxamic acid (SAHA), 5 μg/ml calpeptin (Calp) or combination of SB and Calp, as well as SAHA and Calp, at the same concentrations. B. SKOV-3 ovarian cancer cells were treated with 1 mM SB, 10 μM SAHA, 10 μg/ml Calp or combination of SB and Calp and, also, SAHA and Calp, at the same concentrations. After 48 h, ovarian cancer cells were harvested, suspended in serum-free media and added to the upper chamber. The lower chamber contained conditioned media from untreated control cells. After 12 h, cells were removed from the upper chamber and the membrane was stained and viewed under the microscope. Cell counts were taken from triplicates. Untreated control cell counts were normalized to 100% motility. HDACi, Calp and combination treatment results are shown in percentage motility compared to the untreated control.
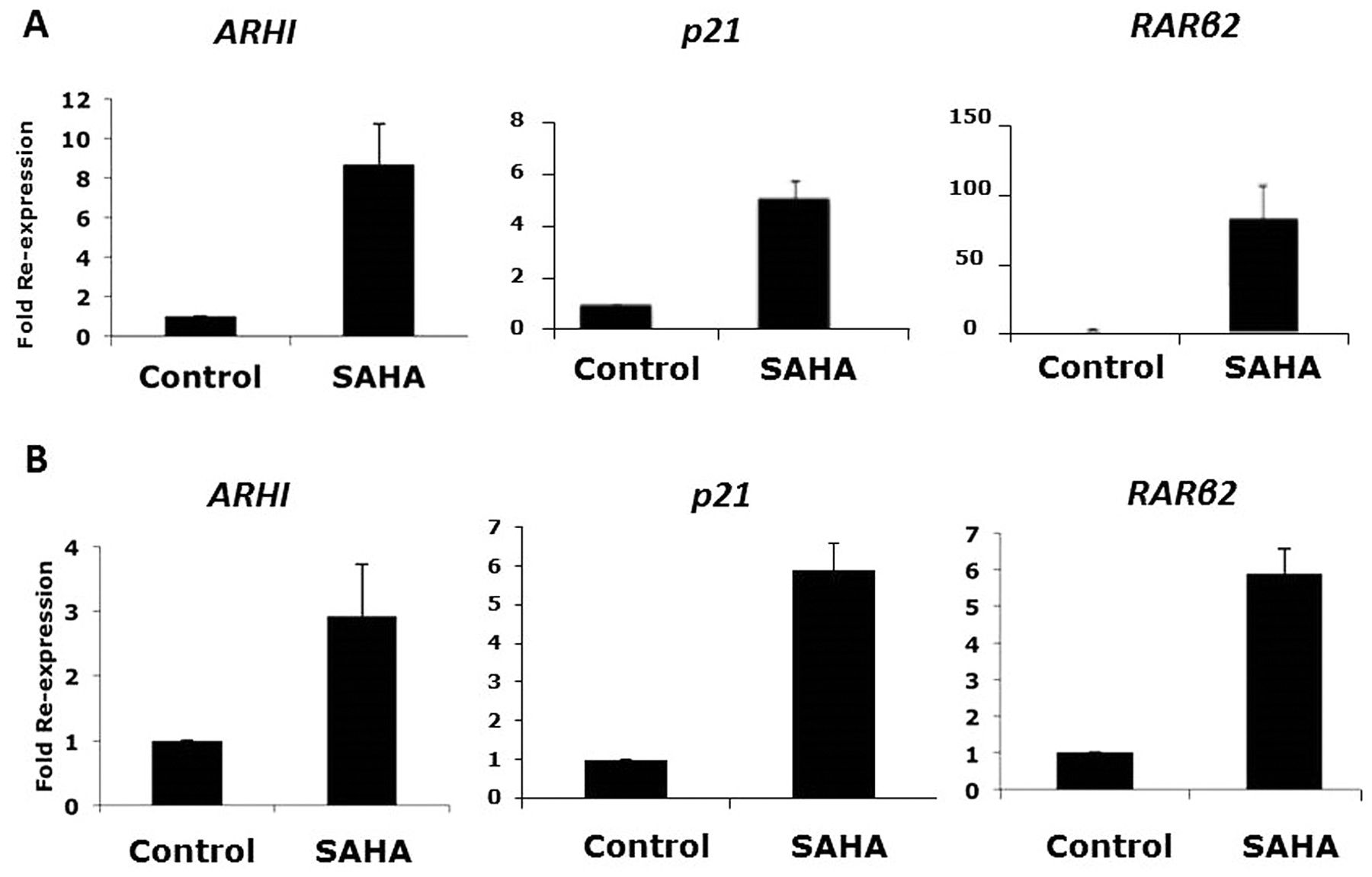
Re-expression of tumor suppressor genes by HDACi. Three TSG (ARHI, p21 and RARβ2) are epigenetically silenced in ovarian cancer cells. To determine the status of these TSG after HDACi, we performed qPCR assays with untreated and treated ovarian cancer cells. SAHA treatment resulted in a 8-fold re-expression of ARHI in CAOV-3 cells and a 3-fold re-expression of ARHI in SKOV-3 cells, with respect to each untreated control cells (Figure 3A and B). A 5-fold increase in p21 re-expression was seen in CAOV-3 cells and an 6-fold increase was seen in SKOV-3 cells (Figure 3A and B). Lastly, RARβ2 re-expression was enhanced 85-fold in CAOV-3 cells and 6-fold in SKOV-3 cells (Figure 3A and B).
The potential cause of ARHI re-expression was further examined by identifying differential methylation at CpG sites across the gene after treatment with HDACi and calpeptin. Five of the sites investigated showed differential methylation levels in both ovarian cancer cells; this demethylation was more prominent in SKOV-3 cells (Table II). Interestingly, calpeptin treatment showed a significant demethylation.
Inhibition of cell cycle, induction of apoptosis and autophagy. The effects of treatments on cell cycle inhibition and apoptosis were then tested. Both CAOV-3 and SKOV-3 displayed similar cell-cycle phase inhibition with the addition of SB, SAHA and calpeptin (Figure 4A and B). CAOV-3 cells treated with a combination of SB or SAHA with calpeptin exhibited a significant 16-18% increase in S-phase arrest (Figure 4A). Similarly, SKOV-3 cells also exhibited S-phase arrest but certain drug treatments portrayed considerably increased inhibition than CAOV-3 cells (Figure 4B). SKOV-3 cells treated with SB, calpeptin and combination of both presented a substantial cell cycle arrest in the S-phase, whereas combination SAHA and calpeptin treatment presented a substantial cell-cycle arrest in the G2/M phase.
In order to determine whether this cell death was caused by the induction of apoptosis or autophagy, both possibilities were investigated. To measure apoptosis, the annexin/PI assay, which provides a comprehensive measurement of all the phases of apoptosis, was utilized (15). The lower right (LR) quadrant depicts cells in the early stage of apoptosis, while the upper right (UR) and upper left (UL) quadrants depict cells in later phases of apoptosis. Both CAOV-3 and SKOV-3 cells exposed to inhibitors exhibit an increase in later phase apoptosis (Figures 4C and D). However, SKOV-3 cells have an overall higher percentage of apoptosis across treatments than CAOV-3 (Figure 4E and F).
Demethylation of ARHI gene.
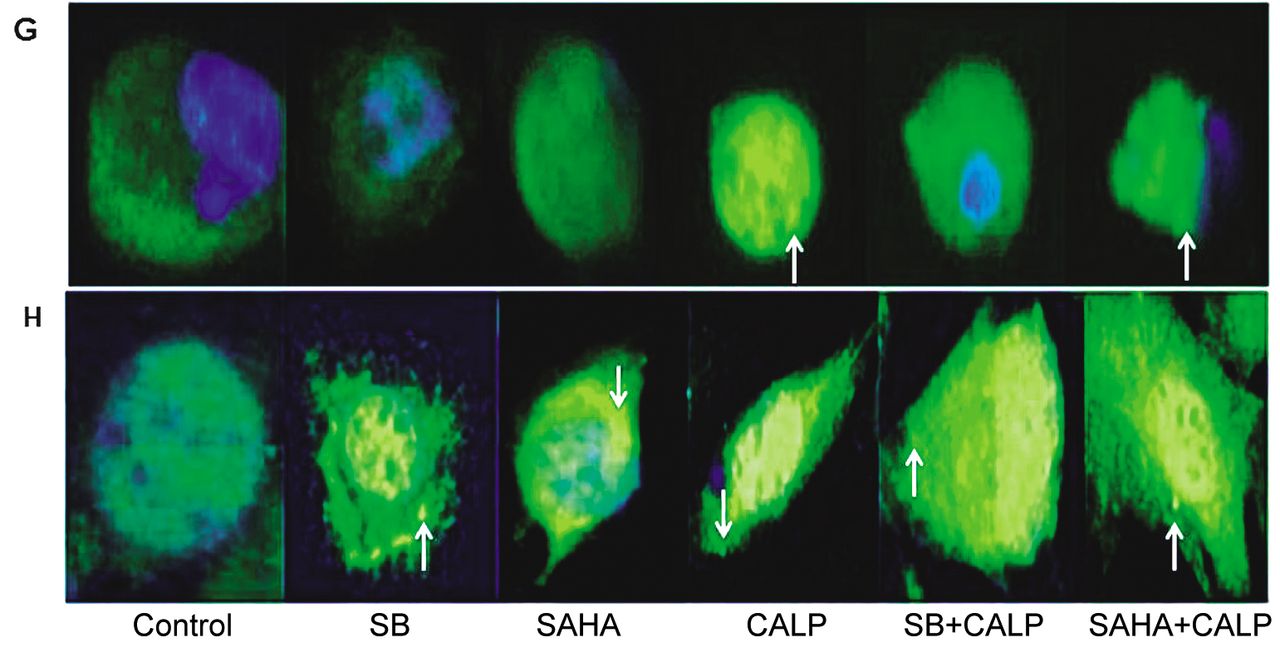
In addition to apoptosis, the morphological changes observed in treated ovarian cancer cells, particularly calpeptin-treated ovarian cancer cells (Figure 1C and D), indicate that they possibly underwent autophagy. To measure autophagy, an immunostaining procedure was used to detect LC3b on phagolysosome membranes. Treatments in both cells produced different patterns of LC3b translocation (Figure 4G and H). CAOV-3 cells treated with a SAHA and calpeptin combination or calpeptin alone showed an increased association between LC3b and phagolysosomes (Figure 4G). In contrast, SKOV-3 cells treated with both HDACi and calpeptin revealed LC3b around phagolysosomes (Figure 4H). Furthermore, SKOV-3 cell shapes were more contorted and elongated than those of CAOV-3. However, in both cell lines, LC3b association was more pronounced in SAHA and calpeptin combination treatments. Untreated control cells showed a diffused staining of lower intensity in the cytoplasm.
Taken together, the results of the cell cycle, apoptosis and autophagy experiments suggest that all of these processes are involved in cell growth inhibition and cell death.
Effects of HDACi and calpeptin on the phosphorylation of ERK and Akt. ERK and Akt pathways are known to have important roles in cancer (8, 10, 15, 23). In order to determine whether HDACi and calpeptin inhibit phosphorylation of ERK and Akt in ovarian cancer cells, a western analysis of each protein lysate was performed. Akt phosphorylation was reduced in CAOV-3 cells when treated with a combination of SB or SAHA with calpeptin, while it was not significantly inhibited in treated SKOV-3 cells (Figure 5A). Similarly, ERK phosphorylation was reduced in CAOV-3 cells when treated with SAHA or combination therapies (Figure 5B). Calpeptin alone did not significantly reduce ERK phosphorylation and SB was not as effective as SAHA. SKOV-3 cells showed similar ERK phosphorylation inhibition patterns (Figure 5B).
Discussion
This study presents results that validate the usefulness of specific epigenetic and cytotoxic drug combination therapies. Specifically, the combination of HDACi with calpain protease inhibitor, calpeptin, was found to enhance growth inhibition in two ovarian cancer cell lines of different characteristics: (i) CAOV-3 (TP53 mutated) and (ii) SKOV-3 (TP53 null and highly metastatic). Two structurally different Class I and Class II HDACi (SB and SAHA, respectively) were employed to ensure that the observed effects were due to the inhibition of HDAC activity and calpeptin was chosen because it regulates signaling pathways involved in cellular growth, apoptosis and autophagy (20) and its inhibition of calpain has anti-thrombotic properties (21, 22). Sub-optimal concentrations, which independently do not inhibit cell growth significantly, were used to demonstrate that greater growth inhibition can only be achieved when HDACi and calpeptin are used in combination (Figure 1A and B). Interestingly, the doses needed for the HDACi and calpeptin combinations to be effective were lower for CAOV-3 than for SKOV-3 ovarian cancer cells. This is consistent with the fact that SKOV-3 cancer cells are more aggressive and resistant and, thus, more difficult to treat. Comparable results in the characteristically different CAOV-3 and SKOV-3 cells suggest that combination therapies may be effective on diverse types of ovarian cancer cell lines and that these effective dosage amounts will vary. Interestingly, the addition of calpeptin further drastically altered the morphology of both types of ovarian cancer cell lines (Figure 1C and D). These results indicated the inclusion of calpeptin-induced cell death.
In addition to uncontrollable cell growth, metastasis also characterizes malignant tumors. The aggressiveness of a tumor is dependent on how motile the individual cancer cells are. Theoretically, the most effective drugs should inhibit growth and promote cell death, as well as inhibit metastasis. Many available chemotherapeutic agents are not effective against all of these characteristics. Some metastasis-inhibiting drugs are not effective at inhibiting cancer cell growth and vice versa. In this study, both HDACi and calpeptin inhibit the motility of CAOV-3 and SKOV-3 ovarian cancer cell lines (Figure 2A and B). Although the individual inhibitors and combinations showed similar effects, we believe that HDACi and calpeptin concentrations used for this experiment were substantial enough to inhibit cell motility even when used as a single agent. Nonetheless, collectively, the results (Figures 1 and 2) show that the combination of HDACi and calpeptin inhibited growth and reduced ovarian cancer cell motility.
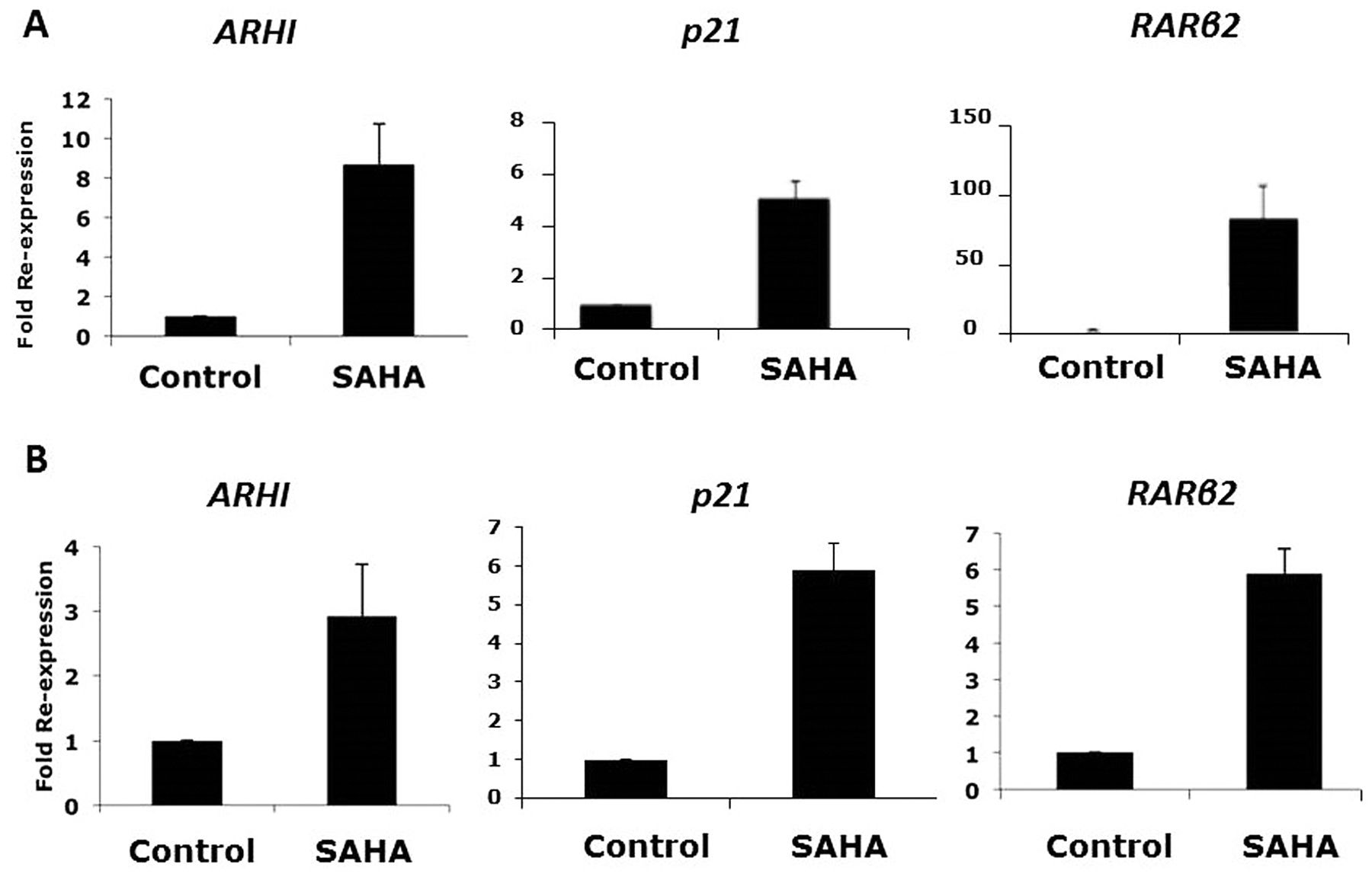
Re-expression of tumor suppressor genes: ARHI, p21, RARβ2. A. CAOV-3 ovarian cancer cells were treated with 10 μM suberanilohydroxamic acid (SAHA) for 48 h. B. SKOV-3 ovarian cancer cells were treated with 10 μM SAHA for 48 h. Total RNA was isolated, cDNA was prepared. qPCR for ARHI, p21 and RARβ2 transcripts were performed. The results were normalized for each sample to the level of β-actin transcripts for each gene in each sample. Average of triplicate data was expressed as a fold re-expression compared to the untreated control cells.
The mechanisms of growth inhibition via drug treatments were further examined. HDACi degrade DNMT1 and, thus, demethylate CpG residues in the upstream region of TSG (10, 15); this study confirms that three such TSG (ARHI, p21 and RARβ2) are re-expressed in both CAOV-3 and SKOV-3 ovarian cancer cell lines after treatment with HDACi SAHA (Figure 3A and B).
These three TSGs are known to be demethylated after HDACi treatment in different types of cancer cells (3, 15) and methylation-specific sequencing of one of candidate gene (ARHI) revealed that this methylation reduction is not uniform across all CpG residues in the gene. Demethylation patterns also varied between the CAOV-3 and SKOV-3 cell types (Table I). Surprisingly, we observed downstream methylation of ARHI in both CAOV-3 and SKOV-3 ovarian cancer cells (+TSS 851 and +TSS 861). Though these regions fall in exon4, they are around the beginning of the transcript. ARHI is specifically expressed in breast and ovarian tissues. Previous studies have shown that methylation in exons regulate transcription of tissue-specific genes (23).
Calpeptin alone was able to reduce methylation since it possibly negatively regulates TET proteins (24). Calpain inhibition sustains TET activity, which may convert 5mC to 5-hydroymethycytosine and ultimately leads to demethylation. In contrast, because the effects of HDACi were different, HDACi may activate yet unidentified DNA demethylases in addition to degrading DNMT1 (10).
The three TSG examined have distinctly different functions. ARHI is an imprinted pro-apoptotic gene, which is similar to RAS in homology, a proto-oncogene, but opposite in function (25). The maternal allele of ARHI is silenced by methylation in normal breast and ovarian tissue and the gene is expressed from the paternal allele. However, in 40% of breast and ovarian cancer cells, the paternal allele is also silenced by methylation, causing loss of heterozygosity (10, 15). P21 is a cell-cycle inhibitor that is usually silenced in many types of cancer cells, including ovarian cancers, which facilitates uncontrolled cell-cycle progression. RARβ2 is a marker of differentiation and is silenced by methylation, since rapid growth of tumor cells is antagonistic to differentiation. Theoretically, re-expression of RARβ2 can limit uncontrolled cell growth, the re-expression of p21 can cause cell-cycle arrest and the re-expression of ARHI can result in induction of apoptosis and autophagy (10, 25). As observed in this research, these TSG are re-expressed in ovarian cancer cells after HDACi treatment, so the effects of these inhibitors on cell cycle progression, apoptosis and autophagy were further assessed.


Cell-cycle inhibition, apoptosis and autophagy. A. CAOV-3 ovarian cancer cells were treated for 48 h with 1 mM sodium butyrate (SB), 10 μM suberanilohydroxamic acid (SAHA) and 10 μg/ml calpeptin (Calp) or combination of SB and Calp, as well as SAHA and Calp, at the same concentrations. B. SKOV-3 ovarian cancer cells were treated for 48 h with 1 mM SB, 10 μM SAHA and 10 μg/ml Calp or combination of SB and Calp and, also, SAHA and Calp, at the same concentrations. The cells were stained with PI and FACS analysis was performed to determine the stages of cell-cycle inhibition in each sample. C. CAOV-3 ovarian cancer cells were treated with 1 mM SB, 10 μM SAHA, 10 μg/ml Calp or combination of SB and Calp, as well as SAHA and Calp, at the same concentrations. D. SKOV-3 ovarian cancer cells were treated with 1 mM SB, 10 μM SAHA, and 10 μg/ml Calp or combination of SB and Calp, as well as SAHA and Calp, at the same concentrations. After a 48-h treatment, the ovarian cancer cells were stained with annexin conjugated to FITC and PI. Apoptosis quantities were measured with flow cytometry and plotted. E. Graph representation of CAOV-3 cancer cell apoptotic percentages from data of C. F. Graph representation of SKOV-3 cancer cell apoptotic percentages from data of D. G. CAOV-3 ovarian cancer cells were treated for 96 h with 0.5 mM SB, 2 μM SAHA, 5 μg/ml Calp or combination of SB and Calp, as well as SAHA and Calp at the same concentrations. The arrow indicates one of the several LC3b-associated phagolysosomes in cells. H. SKOV-3 ovarian cancer cells were treated for 96 h with 1 mM SB, 10 μM SAHA, 10 μg/ml Calp or combination of SB and Calp and, also, SAHA and Calp, at the same concentrations. Next, cells were treated with antibodies followed by DAPI addition and incubated. Cells were viewed and photographed with a fluorescent microscope. The arrow indicates one of the several LC3b-associated phagolysosomes in cells.
The cell-cycle profiles reveal that HDACi in combination with calpeptin arrests the cell division cycle at the S-phase in both CAOV-3 and SKOV-3 ovarian cancer cells, whereas calpeptin alone does not produce much inhibition (Figure 4A and B). Since HDACi re-expresses p21 in both CAOV-3 and SKOV-3 ovarian cancer cell lines, these results were expected. Calpeptin's limited effect on inhibiting cell-cycle progression was expected, as calpain inhibition is customarily linked to induction of apoptosis and autophagy (22).
Because HDACi and calpeptin were successful at inhibiting growth with much lower concentrations in CAOV-3 cells than in SKOV-3 cells (Figure 1A and B), we predicted that CAOV-3 would have increased levels of apoptosis as compared to SKOV-3. However, induction of apoptosis was more apparent in SKOV-3 ovarian cancer cells (Figure 4C-F). Low levels of CAOV-3 apoptosis could be explained by the majority of cells being dead during harvesting and before the assay was performed. This proposition is supported by the fact that the number of live CAOV-3 cells was much lower than that of SKOV-3 cells and most CAOV-3 cells were smaller and rounded in shape, thus suggesting that they had already progressed through cell death (not shown).
In addition, the drastic change in morphology (Figure 1C and D) and the re-expression of ARHI (Figure 3A and B) after treatment with calpeptin alone or in combination with HDACi suggests a possible induction of autophagy. CAOV-3 cells had distinct formations of phagolysosomes, which contained fluorescently stained LC3b, after treatment with calpeptin (Figure 4G and H). HDACi treatment of SKOV-3 cells also showed LC3b on phagolysosomes, although in a lower amount as compared to the combination treatment. Finally, the combination therapy of HDACi and calpeptin resulted in the formation of phagolysosomal complexes with LC3b in both cell types. While a larger signal of LC3b was expected in CAOV-3 cancer cells, the results demonstrate lower numbers of phagolysosomes attached to LC3b compared to that in SKOV-3 cells. This possibility is supported by the fact that lower doses of HDACi and calpeptin in combination produced similar growth inhibition in CAOV-3 cells as compared to SKOV-3 ovarian cancer cells (Figure 1 A and B). Figure 1C and D showed the inhibitors had more drastic effects on CAOV-3 cells than SKOV-3 even when used in the same concentrations. It appears that most of the CAOV-3 cells were already dead, degraded and less in number.

Effects of HDACi and calpeptin on Akt and ERK phosphorylation. A. CAOV-3 ovarian cancer cells (upper left panel) and SKOV-3 ovarian cancer cells (upper right panel) were treated with 1 mM sodium butyrate (SB), 1 μM suberanilohydroxamic acid (SAHA), 20 μg/ml calpeptin (Calp), or combination of SB and Calp, as well as SAHA and Calp, at the same concentrations. Western analysis was performed for Akt phosphorylation (pAkt) in CAOV-3 and SKOV-3 ovarian cancer cells. B. CAOV-3 ovarian cancer cells (lower left panel) and SKOV-3 ovarian cancer cells (lower right panel) were treated with 1 mM SB, 10 μM SAHA, 20 μg/ml calpeptin (Calp) or combination of SB and Calp, as well as SAHA and Calp, at the same concentrations. Western analysis was performed for ERK phosphorylation (pERK) in CAOV-3 and SKOV-3 ovarian cancer cells. Blots were stripped and a western blot followed for Akt and ERK, respectively.

Model of combination therapy using HDACi and calpeptin.
HDACi are known to inhibit DNMTI in cancer cells via inhibition of ERK kinase (10, 17). In this study, SAHA and combination therapies were found to effectively inhibit ERK phosphorylation in both CAOV-3 and SKOV-3 cells, while SB and calpeptin alone were not as effective (Figure 5B). However, the combination treatment showed the inhibition of ERK phosphorylation in both ovarian cancer cell lines. The inhibition of ERK phosphorylation suggests that, in addition to DNMT1 degradation that demethylates TSG and enables their re-expression, these drug combinations also inhibit an imperative pro-growth signaling pathway mediated by ERK kinase.
Additionally, the inhibition of calpain is known to negatively regulate Akt-mediated signaling (22). Akt is an anti-apoptotic kinase and often constitutively phosphorylated in various types of cancer cells. Inhibition of Akt phosphorylation is associated with induction of apoptosis and initiation of autophagy. The inhibition of Akt phosphorylation by calpeptin was not as prominent as hypothesized (Figure 5A). This is likely due the use of sub-optimal doses of calpeptin used in these experiments. Although this low dosage is sufficient at inhibiting cell growth when used in combination with HDACi, a much higher dosage may be required to show complete inhibition of Akt phosphorylation in ovarian cancer cells. This is supported by the fact that the complete inhibition of calpain activity is usually achieved with about 40 μg/ml calpeptin, depending on the cell line used.
This study demonstrates that the combination of HDACi and calpeptin produce enhanced growth inhibition and cell death in different types of ovarian cancer cells. The growth inhibition and cell death result from a combination of cell-cycle arrest and the induction of apoptosis and autophagy that are associated with the demethylation and re-expression of TSG (Figure 6). Additionally, these inhibitors significantly block the motility, as well as associated metastatic potential, of ovarian cancer cells. Although the mechanisms of inhibition are similar between the CAOV-3 and SKOV-3 cell lines, each cell type produced distinct results and required distinct drug doses.
These findings serve as one step towards developing an effective combination therapy for ovarian cancer, which is readily needed given that the current platinum drug treatment often causes resistance and other specific chemotherapies are lacking. The hypothesis that a combination of epigenetic drugs with other cytotoxic drugs may be more effective at treating cancer than either drug alone is not a new idea (5, 11, 13, 15). It is based on the observation that epigenetic drugs promote the re-expression of TSG and this should sensitize cancer cells towards other chemotherapeutic agents (15, 16). Several research groups have already found that HDACi in combination with additional inhibitors are very successful treatments against breast and ovarian cancer cells (3, 10, 15, 16). However, our research group was the first to hypothesize that a combination treatment involving epigenetic drugs is a better choice as it has the potential to inhibit cancer progenitor cell formation, cancer progression and metastasis and, also, to kill cancer progenitor cells and drug-resistant cancer cells to reduce relapse (4, 5, 8-10, 13, 27). We further observed that combination therapies work best when the combined drugs are compatible and increase the efficacy of the each other (unpublished). These specific requirements result in many combinations being unsuccessful. This study begins to explore this complication and reveals how specific drugs that target similar mechanisms can be effective in different ovarian cancer types when used at differential dosages. These improved combination therapies may further be effective in killing cancer progenitor cells and drug-resistant cancer cells, which would help reduce relapse and increase survival of ovarian cancer patients.
Acknowledgements
This study was partially supported by an ACS Grant IRG-72-001-34 to SS. KL, GH, SH and AW were funded by UROP, Boston University. SB and AO were funded by MSSRP, Boston University School of Medicine (BUSM). We thank Dr. Shannon Kokolus Shiel for assistance. We are thankful to Dr. D.V. Faller, Professor at BUSM, for the use of facilities.
Footnotes
This article is freely accessible online.
Conflicts of Interest
The Authors declare no conflict of interest.
- Received August 21, 2016.
- Revision received September 1, 2016.
- Accepted September 2, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}