


Abstract
Background: Previously, we synthesized a new carbocyclic analog of pyrrolo[2,3-d]pyrimidine nucleoside, designated MCS-C3. Recently, we found that LNCaP androgen-responsive prostate cancer cells treated with MCS-C3 rapidly undergo intrinsic apoptosis through dramatic up-regulation of p21CIP1. The present study aimed to evaluate the cellular functions and underlying molecular mechanisms of p21CIP1 on apoptotic induction in LNCaP cells treated with 6 μM MCS-C3. Materials and Methods: Western blots, flow cytometric assay, immunoprecipitation, and transmission electron microscopy analysis were used to measure apoptotic induction in 6-μM MCS-C3-treated LNCaP cells. Effects of MCS-C3 on gene expression of p21CIP1 were measured by semi-quantitative real-time polymerase chain reaction, and small interfering RNA transfection. Results: MCS-C3 induced appreciable caspase-dependent apoptosis associated with the significant up-regulation of p53-dependent p21CIP1 in LNCaP cells. Moreover, this apoptotic induction was caused by direct binding of p21CIP1 to anti-apoptotic B-cell lymphoma 2 (BCL2) protein, and antagonizing BCL2 function. In addition, MCS-C3-mediated apoptotic induction, and up-regulation of p21CIP1 were almost completely blocked by the treatment of androgen-responsive LNCaP cells with flutamide, an androgen receptor (AR) antagonist. Conclusion: We identified that induction of intrinsic apoptosis in LNCaP cells by 6 μM MCS-C3 is associated not only with p53 activation but also with mediation of AR. In the present study, we identified the cellular functions and underlying molecular mechanisms of p53-dependent and AR-associated p21CIP1 on apoptotic induction via direct binding to BCL2 in LNCaP cells treated with 6 μM MCS-C3.
Prostate cancer is the most frequently diagnosed cancer in men and is one of the leading causes of cancer-related fatalities worldwide (1, 2). Prostate cancer, in general, can be treated by surgery, radiation and hormonal therapy (3). Most of all, androgen ablation therapy can trigger death of prostate cancer cells, and remains one of the primary treatment options for all patients with prostate cancer. However, in many cases, patients eventually fail androgen ablation therapy and die due to recurrence of androgen-refractory growth (4).
Uncontrolled cell-cycle progression and apoptotic cell death are the key characteristics of cancer cells due to a homeostatic perturbation between cell proliferation and death. Cyclin-dependent kinases (CDKs) play pivotal roles in cell-cycle progression in cooperation with various endogenous cyclins, and CDK inhibitors, including p21CIP1, p27KIP1, and p57KIP2 (5, 6). In particular, p21CIP1 arrests the cell cycle at the G1 and G2/M phase, and plays a pivotal role as a tumor suppressor, promoting antiproliferative activities. However, despite its important role in arresting cell-cycle progression, recent studies suggest that p21CIP1 also inhibits apoptosis, which might account for its oncogenic effect (7, 8). In fact, p21CIP1 is often overexpressed in a variety of human cancer types and its up-regulation positively correlates with tumor invasiveness and aggressiveness (9-11). Recently, it was reported that p21CIP1 might paradoxically promote apoptosis under certain cellular stresses, such as treatment with an anticancer drug (8). However, the mechanisms involved in p21CIP1-dependent apoptosis are not well understood and seem to be dependent on cell type.
Androgens are steroid hormones that play a pivotal role in the development of prostate cancer and have pleiotropic functions in androgen-responsive cells, such as LNCaP cells (12-15). The cytoplasmic androgen receptor (AR) is a member of the steroid hormone receptor superfamily and can act as a latent transcription factor in response to androgen (1, 3). After binding to androgen, the complex translocates into the nucleus to induce expression of target genes, such as prostate-specific antigen (PSA), CDK2 and p21CIP1, which are involved in many cellular activities, from proliferation to apoptosis (2, 16-19). However, the cellular functions of p21CIP1 mediated by AR on apoptotic induction remain unknown.
Previously, in the course of our screening for novel anticancer compounds, we synthesized a new carbocyclic analog of pyrrolo[2,3-d]pyrimidine nucleoside, designated MCS-C3 (Figure 1A), and reported its anticancer activities in human ovarian cancer PA-1 cells (20). In the present study, we identified the cellular functions and underlying molecular mechanisms of p53-dependent and AR-associated p21CIP1 protein on apoptotic induction in LNCaP androgen-responsive prostate cancer cells treated with 6 μM MCS-C3.
Materials and Methods
Reagents and chemicals. RPMI-1640 medium was purchased from Welgene Fresh Media™ (Gyeonsangbuk-do, Korea). All antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), except for caspase-9 and -3 (Cell Signaling Technology, Beverly, MA, USA) and β-actin (Sigma Aldrich, St. Louis, MO, USA). Control and p53 small interfering RNAs (siRNAs) were purchased from Cell signaling Technology. Amersham™ ECL™ prime western blot detection reagent, protein assay kit, and flutamide were purchased from GE Healthcare (Buckinghamshire, UK), Bio-Rad Laboratories (Hercules, CA, USA), and Sigma Aldrich, respectively.
Cell line and culture. LNCaP human prostate cancer cells were purchased from the American Type Culture Collection (Rockville, NY, USA) and cultured 37°C in a humidified atmosphere of 5% CO2-air using RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) (PAN Biotech, Aidenbach, Germany) and 1% penicillin/streptomycin. The cell density in the culture did not exceed 1×106 cells/ml.
Cell viability assays. CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) was performed to determine the cytotoxicity towards LNCaP cells of MCS-C3 at different concentrations according to the manufacturer's instructions. Briefly, cells (1×104 cells/well) were plated in 96-well plates and treated with 0-100 μM MCS-C3 for 24 h. Then 20 μl of CellTiter 96® AQueous One Solution reagent was added to the wells directly and incubated at 37°C for 4 h. The amount of soluble formazan was measured at 490 nm using a microplate spectrophotometer (Thermo Labsystems Inc, Philadelphia, PA, USA).
Flow cytometric analysis of apoptosis. MCS-C3-treated cells grown in 6-well plates were harvested by trypsin digestion. After washing with ice-cold phosphate-buffered saline (PBS), cells were fixed in 70% ice-cold ethanol and stored at 4°C. For flow cytometric analysis, the cells were incubated with 0.1 mg/ml RNase A for 30 min at 37°C, stained with 50 μg/ml propidium iodide for 30 min on ice, and then measured using a Guava easyCyte™ flow cytometer (Merck Millipore, Darmstadt, Germany).
Western blot analysis. Whole-cell lysates were prepared in protein lysis buffer (50 mM Tris, 5mM EDTA, 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 1 mM sodium o-vanadate, 100 μg/ml phenylmethylsulfonyl fluoride and proteinase inhibitors). The protein content was determined with a Bio-Rad protein assay reagent using bovine serum albumine as the standard. The protein extracts (30 μg) were analyzed using 10-14% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membrane (Millipore Corp., Bedford, MA, USA). The membranes were blocked with 5% (w/v) non-fat dry milk and then incubated with the indicated antibodies in TBS (10 mM Tris–HCl, 150 mM NaCl/pH 7.6) containing 0.1% Tween-20 with gentle shaking for 24 h at 4°C. The secondary antibody was a peroxidase-conjugated goat anti-mouse antibody or goat anti-rabbit antibody. The signals were detected using an ECL western blotting kit and visualized using Chemi-Doc system (Bio-Rad).
Immunoprecipitation. Five hundred micrograms of total protein extract was immunoprecipitated with 5 μg of anti-p21CIP1 or anti-B-cell lymphoma 2 (BCL2) antibody. For co-immunoprecipitation assay Dynabeads® Protein G was used (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Immunoprecipitates were subjected to western blot analysis as described above.
Semi-quantitative real-time polymerase chain reaction (qRT-PCR). Total RNA from cultured cells was isolated by RiboEx™ (GeneAll Biotechnology, Seoul, Korea) according to the manufacturer's instructions. cDNA was prepared from 2 μg total RNA using cDNA kit (Roche Applied Science, Mannheim, Germany) following the manufacturer's instruction. qRT-PCR was performed with IQ™ SYBR Green Supermix (Bio-Rad) on a CFX Connect Real-time system (Bio-Rad). All reactions were assessed for quality by examination of both amplification and dissociation curves. Results were normalized to the level of β-actin (ACTB). The following primers were used in the study: p53 forward primer: CCTCACCATCATCACACTGG; p53 reverse primer: TCTGAGTC AGGCCCTTCTGT; p21CIP1 forward primer: GAGCGATGGA ACTTCGACTT; p21CIP1 reverse primer: CAGGTCCACATGGT CTTCCT; ACTB forward primer: GTGGGGCGCCCCAG GCACCAGGGC; ACTB reverse primer: CTCCTTAATGRCACG CACGATTTTC. The relative expression was calculated using the comparative threshold cycle method, where Ct indicates the fractional cycle number at which the amplified gene amounted to a fixed threshold within the linear phase of amplification. The median Ct of triplicate measurements was used to calculate ΔCt as the Ct for the p53 and p21CIP1 genes. The ΔCt for each sample was compared with the ΔCt for the control, and the difference was expressed as ΔΔCt. The relative quantification was then expressed as the fold-change in p53 and p21CIP1 expression compared with the control condition using the formula 2−ΔΔCt.
siRNA transfection. LNCaP cells were seeded in 6-well plates, two days before transfection. Cells about 80% confluent were transiently transfected with 10 μM of control and p53 siRNA using DharmaFECT® 3 reagent (Thermo Fisher Scientific Dharmacon, CO, USA) according to the manufacturer's protocol.
Transmission electron microscopy (TEM). The cells were pelleted and washed twice with PBS. Fixation was performed with phosphate buffer (pH 7.4) containing 2.5% glutaraldehyde for 3 h at 4°C. The pellets were further incubated in phosphate buffer (0.1 M) with 1% osmium tetroxide. Standard dehydration of the specimen was performed before it was embedded in epoxy resin, sectioned, and stained with paraphenylenediamine. Ultrathin sections were obtained, mounted in copper grids and counterstained with uranyl acetate and lead citrate. The specimens were examined for the molecular characteristics of cell death using an electron microscope (Hitachi H-7600 TEM, Tokyo, Japan).
Statistical analysis. The data are reported in terms of the mean and standard deviation of three independent experiments and were evaluated by Student's t-test. Values of p<0.05 were considered to be statistically significant.
Results and Discussion
MCS-C3-induced apoptosis of LNCaP cells. To identify the effects of MCS-C3 on apoptotic induction in LNCaP androgen-responsive prostate cancer cells, we investigated cell viability using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, morphological characteristics by TEM analysis, and the expression pattern of apoptosis-related proteins by western blotting in LNCaP cells treated with 0 and 6 μM MCS-C3 for 24 h.
The results of the MTT assay demonstrated that the proliferation of MCS-C3-treated LNCaP cells was not inhibited in a dose-dependent manner (Figure 1B). The maximal anti-proliferative effect was detected at a specific concentration of MCS-C3, namely 6 μM (calc. half-maximal inhibitory concentration value=5.2 μM). Interestingly, the viability of LNCaP cells treated with concentrations lower (3 μM) and higher (20 μM) than 6 μM was not significantly inhibited.
In this context, we performed TEM analysis to identify the morphological changes of MCS-C3-treated LNCaP cells. The morphological characteristics of apoptotic cells, such as chromatin condensation and the formation of crescent-shaped bodies, were clearly found in 6-μM-treated LNCaP cells (Figure 1C).
To determine whether apoptosis-associated proteins are involved in the mediation of MCS-C3-induced apoptosis, we examined activation of caspases and the cleavage of poly(ADP-ribose) polymerase (PARP) in LNCaP cells treated with 6 μM MCS-C3 by western blot analysis. The proteolytic cleavage of PARP, an enzyme involved in DNA repair, and activation of procaspases into caspases were identified in 6-μM-treated LNCaP cells (Figure 1D). In general, the cleavage of PARP has been used as a typical indicator of apoptotic induction in response to drug treatment (21, 22).
Furthermore, the expression of anti-apoptotic BCL2 protein significantly decreased in LNCaP cells treated with 6 μM MCS-C3. This down-regulation of BCL2 may be associated with the strong up-regulation of p21CIP1 in LNCaP cells treated with 6 μM MCS-C3 (Figure 1D). The BCL2 protein plays an important role in determining cell survival and death through regulation of the re-distribution of mitochondrial cytochrome c to the cytosol (23, 24). Therefore, in order to examine whether mitochondria-mediated intrinsic apoptosis is involved in MCS-C3-induced apoptosis, we analyzed the cytochrome c release from mitochondria to the cytosol using western blotting. Cytochrome c release into the cytosol was observed in LNCaP cells treated with 6 μM MCS-C3 for 24 h (Figure 1E). These findings demonstrate that appreciable caspase-dependent intrinsic apoptosis was apparently induced in LNCaP cells treated only with 6 μM MCS-C3 for 24 h.
Recently, considerable attention has been paid on the cellular function of p21CIP1, whose anti- or pro-apoptotic functions involved in either tumorigenesis or tumor suppression depend on cell type and cellular conditions (8). However, the underlying mechanisms involved in p21CIP1-dependent apoptotic induction are not well-understood.
In our experiment, the expression of p21CIP1 dramatically increased in apoptotic LNCaP cells treated with 6 μM MCS-C3. Concerning the p21CIP1-dependent mechanism of the apoptotic induction, our results suggest the molecular mechanism of p21CIP1 may be by its acting as an apoptosis inducer or a tumor suppressor.
MCS-C3-induced apoptosis through p53-mediated up-regulation of p21CIP1. p21CIP1, a well-known CDK inhibitor and one of the transcriptional targets of p53, is a protein up-regulated after cellular stress stimuli through classical p53-dependent activation (25, 26). To determine whether the up-regulation of p21CIP1 caused by MCS-C3 in LNCaP cells is associated with active p53 as an up-stream transcription factor, we examined p21CIP1 expression when p53 transcription was knocked-down by transfection with p53 siRNA.
Analysis using qRT-PCR demonstrated that p21CIP1 expression in LNCaP cells was dramatically up-regulated by more than 20-fold on treatment with 6 μM MCS-C3, whereas p53 knock-down reduced the mRNA level of p21CIP1 in cells treated with 6 μM MCS-C3 by approximately 91% (Figure 2A). Thus, MCS-C3-induced p21CIP1 up-regulation is apparently associated with p53 activation in LNCaP cells.
Furthermore, knock-down of p53 clearly down-regulated not only the protein level of p21CIP1 but also the cleavage of PARP in LNCaP cells treated with 6 μM MCS-C3 (Figure 2B). In addition, flow cytometric analysis demonstrated that the distribution of the cell population in the apoptotic (sub-G1) phase apparently decreased in p53 knocked-down LNCaP cells treated with 6 μM (Figure 2C). Therefore, we can conclude that MCS-C3-induced apoptosis in LNCaP cells is obviously associated with p53-mediated up-regulation of p21CIP1.

MCS-C3 induces apoptotic cell death in LNCaP androgen-dependent prostate cancer cells at 6 μM. A: The chemical structure of MCS-C3, 4-amino-6-bromo-1-cyclopentyl-1H-pyrrolo[2,3-d]pyrimidine-5-carboxamide. B: LNCaP cells treated with MCS-C3 at the indicated concentrations for 24 h were quantified for cell viability by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. **p<0.01, compared with control. C: Morphological analysis by transmission electron microscopy of LNCaP cells treated with MCS-C3 at the indicated concentration for 24 h (scale bar=2 μm). D: The expression of apoptosis-associated proteins and p21CIP1. E: The levels of cytochrome c release in the cytosol from mitochondria were analyzed by western blotting in 6 μM-treated LNCaP cells. β-actin was used as a loading control. BCL2: B-cell lymphoma 2; PARP: Poly (ADP-ribose) polymerase.
However, further investigation remains to be performed in order to clarify the discrepancy between strong suppression (ca. 91%) of p21CIP1 expression and relatively weak attenuation (ca. 50%) of apoptosis in p53 knocked-down LNCaP cells treated with 6 μM MCS-C3 as shown in Figure 2. A considerable level of p21CIP1 still existed despite the significant reduction of p53 expression (Figure 2B). Thus, we speculate that this residual p21CIP1 might be produced by association of AR. In fact, several recent studies indicated that transcription of p21CIP1 is involved in binding of AR to androgen response elements in p21CIP1 promoter in androgen-responsive prostate cancer cells (27, 28).
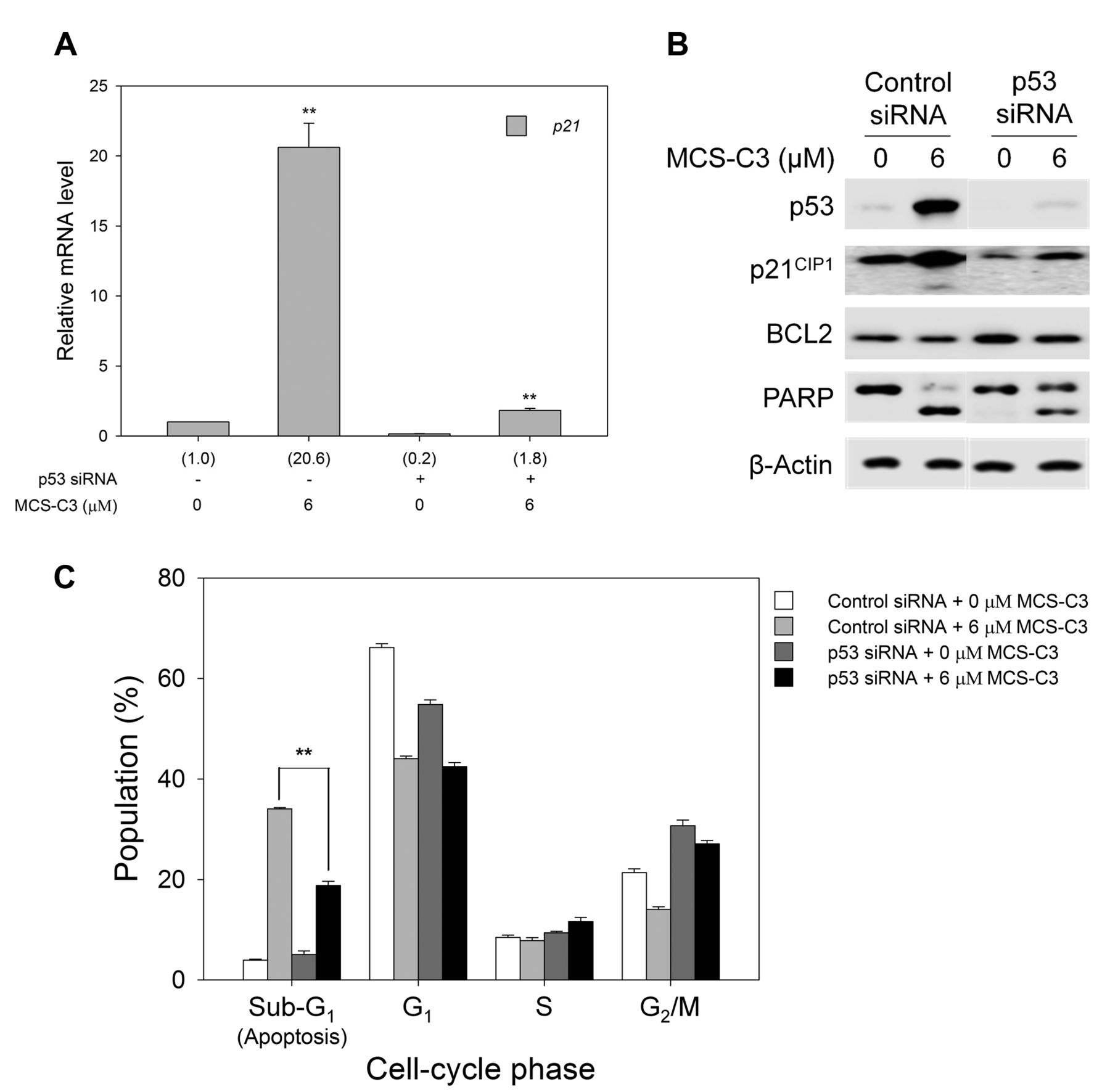
The p53-dependent up-regulation of p21CIP1 is associated with induction of apoptosis in MCS-C3-treated LNCaP cells. Expression of p53 was knocked down by transfection with small interfering RNA. Then the cells were treated with 6 μM MCS-C3 for 24 h. A: Relative mRNA levels of p21CIP1 were determined by semiquantitative real-time polymerase chain reaction. B: The expression of p21CIP1 and BCL2 at the protein level was analyzed by western blotting. C: The distribution of the cell-cycle populations was examined using flow cytometry. **p<0.01, Control siRNA + 6 μM MCS-C3 vs. p53siRNA + 6 μM MCS-C3. BCL2: B-cell lymphoma 2; PARP: Poly (ADP-ribose) polymerase.
AR-associated expression of pro-apoptotic p21CIP1. As described above, several studies suggest that AR-associated expression of p21CIP1 may involve an anti-apoptotic function in prostate cancer cells. However, the cellular functions of AR-mediated p21CIP1 on apoptotic induction remain still unknown. Thus, further investigation should be performed to characterize the hypothetical AR-associated p21CIP1 expression that would induce apoptosis in LNCaP androgen-responsive and androgen-dependent prostate cancer cells.

The androgen receptor (AR) regulates apoptosis via p21CIP1 expression in LNCaP cells treated with 6 μM MCS-C3. A: Effect of flutamide, an AR antagonist, on expression of p21CIP1 and other apoptosis-associated proteins was determined by western blotting. B: The distributional changes of cell-cycle populations after flutamide treatment were measured by flow cytometric analysis. **p<0.01, compared with control. ##p<0.01, 6 μM MCS-C3 vs. 6 μM MCS-C3 + flutamide 100 μM. BCL2: B-cell lymphoma 2; PARP: Poly (ADP-ribose) polymerase.
Previously, we investigated the effects of MCS-C3 on apoptotic induction and the up-regulation of p21CIP1 was never detected in DU145 and PC3 cells, prostate cancer cells not responsive to androgen (data not shown). Moreover, the effects of MCS-C3 on cell proliferation in these cells treated with MCS-C3 were never identified. As such, AR generally plays an important role in androgen-responsive prostate cancer cell proliferation.
To determine whether the apoptotic induction in MCS-C3-treated LNCaP cells is mediated by signaling pathways involved in AR, an AR inhibitor, flutamide, was used to antagonize AR activity in LNCaP cells treated with 6 μM MCS-C3. Western blot analysis demonstrated that inhibition of AR activity significantly reduced the expression of p21CIP1 and also suppressed apoptotic induction, reducing activation of caspase and PARP cleavage (Figure 3A). In addition, flow cytometric analysis also revealed that pre-treatment of cells with 100 μM flutamide significantly reduced the apoptotic cell population (sub-G1) phase for LNCaP cells treated with 6 μM MCS-C3 (Figure 3B).

p21CIP1 directly binds to B-cell lymphoma 2 (BCL2). A: Localization and increased expression of p21CIP1 and BCL2 proteins in the mitochondrial and cytosolic fractions of LNCaP cells treated with 6 μM MCS-C3 were analyzed by western blotting. B: Protein–protein interactions of p21CIP1 and BCL2 protein in LNCaP cells treated with 6 μM MCS-C3 for 0, 9, and 15 h was detected by immunoprecipitation (IP) using anti-p21CIP1 or anti-BCL2 antibody. BCL2: B-cell lymphoma 2.
Therefore, we conclude that apoptosis of LNCaP cells induced by 6 μM MCS-C3 is associated with hypothetical AR-mediated p21CIP1 expression that would apparently induce intrinsic apoptosis in LNCaP androgen-responsive and androgen-dependent prostate cancer cells.
Protein–protein interaction of p21CIP1 with anti-apoptotic BCL2 protein. BCL2 protein localized in mitochondria prevents the apoptosis-associated release of cytochrome c and apoptosis-inducing factors from the mitochondrial inter-membrane space into the cytoplasm (29). It was reported that down-regulation of p21CIP1 may be associated with the up-regulation of BCL2 in breast cancer cells (30).
To investigate whether the effect of MCS-C3 on apoptotic induction involves the possible interaction of p21CIP1 with anti-apoptotic BCL2 protein, we examined the cellular localization of p21CIP1 and BCL2 in LNCaP cells treated with 6 μM MCS-C3. Western blot analysis demonstrated that the level of p21CIP1 was increased both in the mitochondrial and the cytosolic fraction after 6 μM treatment, indicating that p21CIP1 is predominantly localized in mitochondria. In contrast, the level of BCL2 remained the same with a small reduction, if any, in mitochondria, but apparently decreased in the cytosol as a consequence of MCS-C3 treatment (Figure 4A).The results of immunoprecipitation assay revealed the evident protein–protein interaction between p21CIP1 and BCL2 protein in LNCaP cells treated with 6 μM MCS-C3 for 15 h (Figure 4B). These results support a molecular mechanism for MCS-C3-induced apoptosis in which p21CIP1 neutralizes the anti-apoptotic outcome of BCL2 by blocking BCL2 function via protein–protein interaction. This result clearly suggests that p21CIP1, an apoptosis inducer, could functionally compete with BCL2 protein, an anti-apoptotic factor.
In conclusion, we found that induction of intrinsic apoptosis in LNCaP cells induced by 6 μM MCS-C3 is associated not only with p53 activation but also with mediation of AR. Moreover, we identified the cellular functions and underlying molecular mechanisms of p53-dependent and AR-associated p21CIP1 expression in apoptotic induction via direct binding to BCL2 in LNCaP androgen-responsive prostate cancer cells treated with 6 μM MCS-C3. For further investigation, we will explore the mechanism of apoptotic induction in LNCaPp21−/− cells and LNCaP subline cells with different androgen-responsiveness and dependency, such as LNCaPE9 and LNCaPC4−2 cells.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (2013R1A1A2011531).
Footnotes
↵* These Authors contributed equally to this work.
Conflicts of Interest
The Authors declare no conflicts of interest with regard to this study.
- Received October 23, 2015.
- Revision received November 26, 2015.
- Accepted December 2, 2015.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.