


Abstract
Background: We evaluated the ability of itraconazole to enhance the effects of bevacizumab in bevacizumab-resistant cancer cells, endothelial cells, and cancer-associated fibroblasts (CAFs). Materials and Methods: Human gastrointestinal cancer cell lines (HT-29, MKN-28 and MKN-45), human umbilical vein endothelial cells (HUVECs), and CAFs established from human colon cancer were used. In each of these cell lines, cell growth, apoptosis, and angiogenesis were evaluated with bevacizumab with and without itraconazole both in vitro and in vivo. Results: Itraconazole suppressed HUVEC growth by apoptosis through inhibition of mitogen-activated protein kinase and ribosomal protein S6 kinase signaling. Itraconazole also suppressed monocyte chemoattractant protein-1 secretion and the growth of CAFs. In xenografts, compared to monotherapy with either agent alone, combined treatment with itraconazole and bevacizumab significantly reduced tumor volume, tumor weight, and microvessel density. Conclusion: Itraconazole-dependent suppression of endothelial cell and CAF growth resulted in synergistic effects with bevacizumab in bevacizumab-resistant cancer cells.
Bevacitumab (Avastin) is a monoclonal antibody against vascular endothelial growth factor (VEGF) commonly used as an anti-angiogenic drug in the treatment of colorectal, renal (1) and breast cancer. Several studies have reported that bevacizumab enhances the anticancer effects of other chemotherapeutic drugs when used in combination treatments (2). VEGF is the most potent angiogenic factor released from cancer cells (3); therefore, inhibition of VEGF receptor signaling via bevacizumab-dependent neutralization of VEGF ligand on endothelial cells causes a decrease in proliferation, mitosis, and tubule formation. However, the exact details of this anticancer mechanism are still unclear. Moreover, different types of cancer exhibit different responses to bevacizumab, and resistance to bevacizumab is well documented. Biomarkers that can be used to predict the efficacy of bevacizumab have still not been identified (4). While almost all colorectal and lung carcinomas exhibit sensitivity to bevacizumab treatment, the sensitivity of most gastric carcinomas to this drug are poor. The reasons for this poor response of gastric cancer are still unclear.
In addition to inherent resistance to bevacizumab depending on cell type, almost all tumors develop resistance to the antitumor effects of bevacizumab after its continuous clinical use, despite adequate initial response to treatment (5-9). Studies have suggested that this decrease in sensitivity is not due to a VEGF-dependent mechanism but is instead mediated by an alternative angiogenic pathway. Moreover, this resistance to bevacizumab appears to occur through several mechanisms that vary depending on cell type (7-11). The mechanisms of bevacizumab resistance are thought to be caused by the interaction between cancer and stromal cells, called the cancer-stromal interaction (CSI). However, the CSI is a multifaceted effect involving multiple signaling pathways. To overcome this resistance, it is necessary to block not one particular mechanism but either the mechanisms specifically occurring in each case, or all the mechanisms of bevacizumab resistance.
Recently, itraconazole, an anti-fungal drug, was reported to have anti-angiogenic effects. In some benign diseases, the use of itraconazole has been reported to improve hemorrhage status. Furthermore, some clinical trials involving the use of itraconazole in cancer therapy have been initiated (12, 13). However, the precise role and effects of itraconazole treatment in cancer are unknown.
In the present study, we sought to evaluate the anti-angiogenic mechanisms of itraconazole compared to those of bevacizumab. Moreover, we investigated the possible synergistc anti-angiogenic and antitumor effects of combined treatment with itraconazole and bevacizumab in bevacizumab-resistant cancer cell lines in vitro and in vivo.
Materials and Methods
Isolation and culture of human colon fibroblasts. We established a fibroblast cell line from a specimen resected from a 64-year-old Japanese male patient with well-differentiated colon cancer. The technical procedure was similar to that described in a previous report (14). After receiving written informed consent, colon carcinoma tissue and nonmalignant colon tissue were collected. After fragmenting with scissors, the tissues were incubated in Dulbecco's modified Eagle's medium (DMEM) containing 1000 U/ml dispase (Godo Shusei, Tokyo, Japan) for 2 h and were then cultivated in DMEM containing 5% fetal bovine serum (FBS) and 1% antibiotic-antimycotic solution at 37°C in an atmosphere containing 5% CO2. To exclude the possibility that epithelial cells were included, cultivated cells were immunostained with vimentin, and we confirmed that all the cells were positive for vimentin expression. Cancer-associated fibroblasts (CAFs) were defined as those cells that were positive for α-smooth muscle actin (SMA). CAFs were used between passages 3 and 6.
Cell-growth assay. MKN-28 cells, MKN-45 cells, HT-29 cells, CAFs, and human umbilical vein endothelial cells (HUVECs) (Lonza, Basel, Switzerland) were treated with increasing concentrations of itraconazole (Janssen Pharmaceutical, Tokyo, Japan) (100 nM, 1 μM, 10 μM and 100 μM) on days 1 and 3. The number of viable cells was measured in triplicate on day 4 using WST-1 assays (TaKaRa BIO, Shiga, Japan). HUVEC proliferation was also evaluated in response to treatment with bevacizumab (Chugai Pharmaceutical, Tokyo, Japan) (0.001, 0.01, 0.1, 1, and 10 μg/ml) using the same procedure. In order to determine whether the anti-angiogenic mechanisms of these agents were mediated through VEGF, we evaluated the proliferation of HUVECs in the presence of different concentrations of VEGF (Abcam, Cambridge, MA, USA) (1, 5, 10, and 50 ng/ml) with bevacizumab or itraconazole.
Flow cytometry. Annexin assays and evaluation of cell-cycle distribution were carried out using flow cytometry on a FACSCalibur instrument (Becton-Dickinson Biosciences, San Diego, CA, USA). HUVECs were plated in 6-cm plastic dishes (BD Falcon, Franklin Lakes, NJ, USA) in DMEM supplemented with 10% FBS on day 0. On days 1 and 3, HUVECs were treated with 100 nM itraconazole or 100 μg/ml bevacizumab or saline as control. Annexin assays were then carried out using a fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit (BD Biosciences). For cell-cycle analysis, harvested and washed HUVECs were analyzed using a CycleTEST PLUS kit (Becton Dickinson) according to the manufacturer's protocol.
Angiogenesis assay. We established an angiogenesis model using colon fibroblasts and HUVECs. On day 0, 3×104 fibroblast cells were seeded in each well of a 24-well plate in DMEM containing 5% FBS. After overnight incubation (day 1), 3×103 HUVECs were also seeded in each well. Two wells were used for each condition. On days 2, 5, and 8, cells were treated with bevacizumab (5 ng/ml) or itraconazole (100 nM). On day 11, cells were fixed and immunostained with antibodies to cluster of differentiation 31 (CD31) (Abcam, Cambridge, MA, USA) to compare the vascular area and the number of vascular joints were counted as an indicator of vascular network formation. Images taken at ×40 magnification were compared.
Western blotting. To evaluate the efficacy of itraconazole as an inhibitor of mammalian target of rapamycin (mTOR), HUVECs were seeded in 6-cm dishes then exposed to 0, 1 μM, or 100 nM itraconazole for 24 hours. We then used PathScan Multiplex Western Cocktail I (Cell Signaling, Danvers, MA, USA) for evaluation of the phosphorylation of 90 ribosomal s6 kinase (90RSK), protein kinase B (AKT), p44/42 mitogen-activated protein kinase (MAPK) and S6 by semi-quantitative western blotting. RAB11 was used as reference protein.
Cytokine secretion from cancer cells. To evaluate the effects of itraconazole on the secretion of angiogenic cytokines from cancer cells, we analyzed specific proteins from MKN-28 cells using a Human Angiogenesis Antibody Array C series 1000 (RayBiotech, Norcross, GA, USA). This kit can be used to evaluate 43 angiogenic cytokines, including interleukin (IL)-1α and β, IL2, IL4, IL6, IL8, VEGF, VEGFD, and insulin-like growth factor (IGF)-1. After incubating MKN-28 cells with 100 nM of itraconazole or saline as control, culture supernatants were collected, and cytokine secretion was measured according to the instructions provided with the array kit.
Cytokine secretion from CAFs. To evaluate the effects of itraconazole on angiogenic cytokines, we analyzed specific proteins from CAFs using a Proteome Profiler Array Human Angiogenesis Array Kit (R&D Systems Inc., Minneapolis, MN, USA). VEGF, VEGFC, vasohibin, phosphatidylinositol-glycan biosynthesis class F protein (PIGF), platelet derived growth factor (PDGF), urokinase plasminogen activator (uPA), monocyte chemoattractant protein (MCP), and IL1β were evaluated with this assay. Fibroblasts and cancer cell lines were seeded into 6-well plates at a density of 1×105 cells/ml. Following overnight incubation, cells were treated with 0 or 100 nM itraconzaole. After 48 h, culture supernatants were collected, centrifuged to pellet any detached cells, and analyzed according to the instructions provided with the array kit.
Animal experiments. Growing cancer cells (5×106 cells, MKN-28 and HT-29) were injected subcutaneously into the left abdominal flanks of five- to 6-week-old male athymic nude mice of the KSN strain with 30g body weight purchased from Japan SLC (Hamamatsu, Japan).
When the subcutaneous tumors developed to approximately 8 mm in maximal diameter, mice were treated with bevacizumab [1 mg/mouse, intraperitoneal (i.p.) injection twice a week], itraconazole (500 IU/mouse, i.p. injection daily), a combination of bevacizumab plus itraconazole at the above concentrations, or vehicle (400 μl mouse, i.p. injection twice a week) for 4–5 weeks (6 mice/group). Maximum tumor diameter (L) and diameter perpendicular to that axis (W) were measured twice a week. Tumor volume was estimated by the following formula: L×W2×1/2. Mice were sacrificed after 5 weeks of treatment Subcutaneous tumors were then removed and weighed. Collected tumors were used for immunohistochemical observations and RNA extraction. Tumors were fixed in formalin-free zinc fixative (BD Biosciences Pharmingen, San Diego, CA, USA) for 24 h, embedded in paraffin, and sectioned at a thickness of 4 μm. Immunohistochemical staining for blood vessels was carried out by the indirect immunoperoxidase method as described above using a rat monoclonal antibody to mouse CD31 (BD Biosciences Pharmingen) as the primary antibody. The microvessel density of each tumor was determined by light microscopy. The number of structures positive for CD31 expression in the densest vascular field in each tumor was counted at a magnification of ×100 (15).

Analysis of cell proliferation and angiogenesis following treatment with itraconazole or bevacizumab. Cell proliferation was measured in response to different concentrations of itraconazole and bevacizumab in HT-29 cells (A), MKN-28 cells (B), cancer-associated fibroblasts (CAFs) (C), and human umbilical vein endothelial cells (HUVECs) (D, E). The proliferation of HUVECs was also measured in the presence of different concentrations of vascular endothelial growth factor (VEGF) alone (F) or in combination with (G) 10 μg/mL bevacizumab or (H) 1 μM itraconazole. Proliferation is plotted as the percentage relative to the number of cells in the control untreated sample. Bars indicate the mean±SEM.
Statistical analysis. Student's t-test was used to evaluate statistical differences between groups. Significant differences were considered as p<0.05.
Results
Effects of bevacizumab and itraconazole on proliferation of cancer cells, HUVECs, and CAFs. Neither itraconazole nor bevacizumab inhibited the proliferation of MKN-28 and HT-29 cells in vitro, even when used at the highest concentration. Moreover, while bevacizumab had no significant effect on the growth of CAFs or HUVECs, itraconazole significantly inhibited the growth of both of these cell lines. The half-inhibitory concentration (IC50) of itraconazole in HUVECs was 5 μM, and almost all HUVECs died when treated with 1 μM itraconazole. In the CAF cell line, the IC50 of itraconazole was 1 μM (Figure 1).

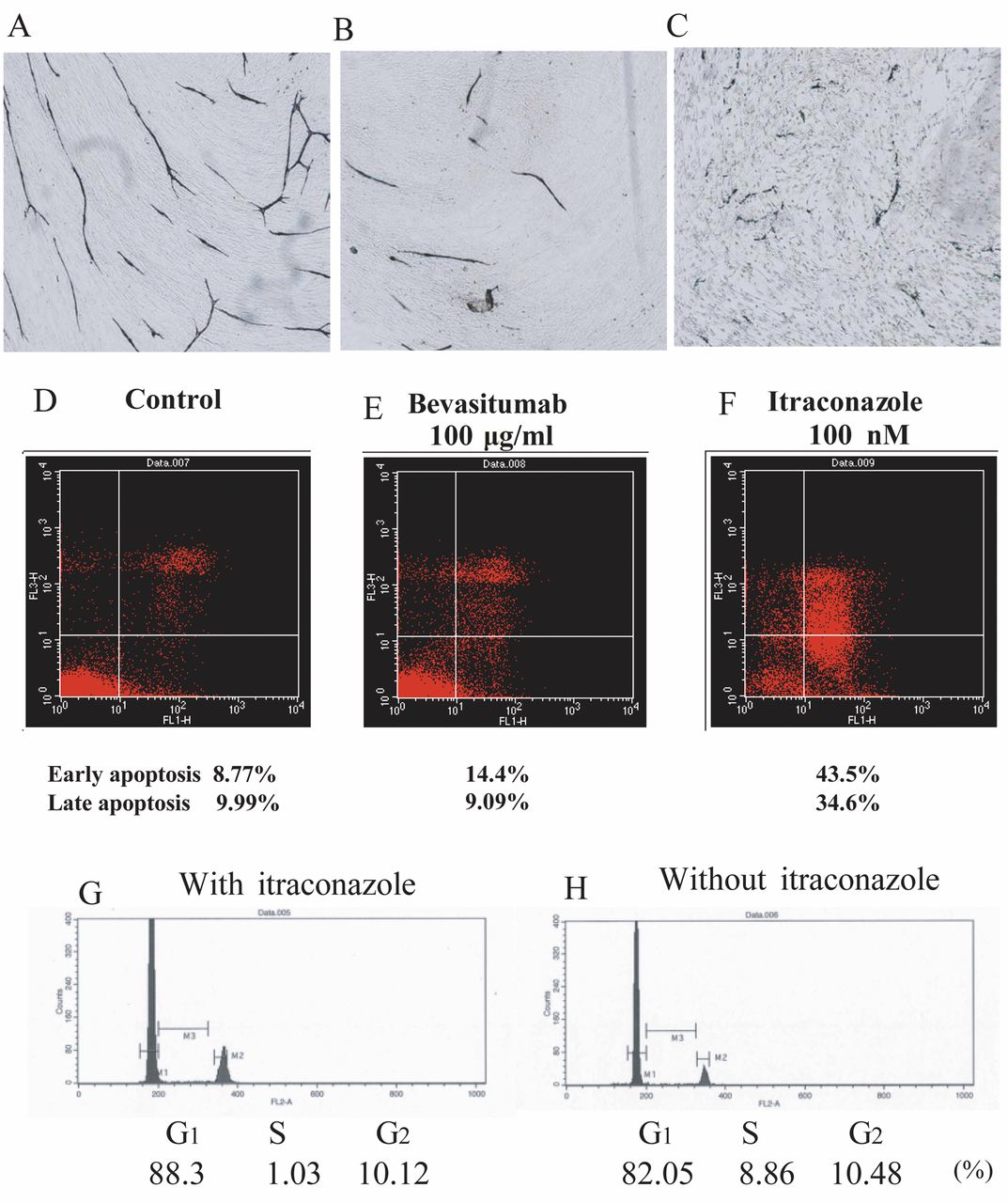
Induction of apoptosis by itraconazole. Upper panels: Representative images of angiogenesis assays following treatment of human umbilical vein endothelial cells (HUVECs) with bevacizumab or itraconazole. HUVECs in the control (A), after treatment with 100 μg/mL bevacizumab (B), and after treatment with 1 μM itraconazole (C). Middle and lower panels: Annexin assays were performed in the control (D) and in cells (E) treated with bevacizumab or itraconazole (F) to assess the induction of apoptosis. The cell cycle distribution was also measured by flow cytometry in HUVECStreated with itraconazole (G) or left untreated (control) (H).

Phosphorylation of ribosomal S6 kinase (RSK), mitogen-activated protein kinase (MAPK), and S6 in human umbilical vein endothelial cells (HUVECs) and analysis of angiogenic factors secreted by MKN-28 cells and cancer-associated fibroblasts (CAFs) with or without itraconazole in vitro using proteome arrays. HUVECs (A) were treated with 1 or 100 μM itraconazole, and the phosphorylation of p44/42 MAPK, p90RSK, Akt, and S6 was analyzed by western blotting. A protein molecular weight marker was included in the first lane. MKN-28 cells (B): MKN-28 secreted interleukin-8 (IL-8), tissue inhibitors of metalloproteinase (TIMP)-1, TIMP-2, chemokine (C-X-C motif) ligand (CXCL)13, and urokinase plasminogen activator receptor (uPAR), which may be associated with bevacizumab resistance in MKN-28 cells. vascular endothelial growth factor (VEGF) (box 6) was not detected. Itraconazole did not influence the secretion of VEGF by MKN-28 cells. Boxes: 1, CXCL13; 2, IL-8; 3, TIMP-1; 4, TIMP-2; and 5, uPAR. The other spots in the image are positive controls. CAFs (C): Representative images of Proteosome Profiler Assay membanes probed with conditioned medium collected from itraconazole- or saline-treated cells. A: Positive control, B: negative control, C: plasminogen activator inhibitor-1, D: TIMP-4, E: IL-1β, F: VEGF, and G: monocyte chemoattractant protein 1 (MCP-1). Itraconazole treatment dramatically suppressed the secretion of MCP-1 (D).

Effects of bevacizumab and itraconazole on xenograft tumors in vivo. Xenograft tumors were established from HT-29, MKN-28, and MKN-45 cells, and mice bearing these tumors were treated with bevacizumab (BV), itraconazole (ITZ), or a combination of both agents for 5 weeks. A: Volume of HT-29 tumors, B: weight of HT-29 tumors, C: volume of MKN-28 tumors, and D: weight of MKN-28 tumors. E: Representative images of microvessels in control and treated MKN-28-derived tumors in mice. F: Microvessel density assays were performed as described in the Materials and Methods, and a representative graph of the data is shown. NS: Not significant.
Next, we evaluated the angiogenic effects of VEGF on HUVECs in the presence of bevacizumab (10 μg/ml) or itraconazole (1 μM). While VEGF promoted HUVEC growth in a dose-dependent manner, bevacizumab inhibited the proliferation of HUVECs, regardless of the presence of VEGF. However, VEGF stimulated HUVEC growth, even in the presence of 1 μM itraconazole. This indicates that the anti-angiogenic mechanism of itraconazole is not dependent on the inhibition of VEGF signaling in HUVECs.
Angiogenesis assay. To examine the effects of bevacizumab and itraconazole on angiogenesis, we performed angiogenesis assays on HUVECs. As shown in Figure 2A-C, HUVECs in the control group grew and branched after 11 days of culture. Treatment with bevacizumab reduced HUVEC growth and branching, thereby markedly reducing the vascular area, number of joints, and length of tubules compared to those of the control. No structural disturbances or fibroblast disruption was observed in bevacizumab-treated cells. Itraconazole elicited the same effects on the vascular area and network formation as bevacizumab, but also induced apoptotic changes in the remaining HUVECs. In addition, itraconazole treatment resulted in the death of fibroblasts.
Effects of bevacizumab and itraconazole on apoptosis and the cell cycle, as measured by FACS. Annexin assays were performed to measure the effects of itraconazole on apoptosis in HUVECs (Figure 2D-F). In control cells, the percentages of cells in early and late apoptosis were 8.8% and 10.0%, respectively; treatment with itraconazole significantly increased these rates to 43.5% and 34.6%, respectively. On the other hand, cells treated with bevacizumab did not exhibit differences in the rates of early and late apoptosis compared to the control, at 14.4% and 9.1%, respectively. Thus, these results demonstrate that itraconazole, but not bevacizumab, induces a significant increase in both early and late apoptosis.
To evaluate the effects of itraconazole and bevacizumab on cell progression, we analyzed the cell-cycle distribution as described in the Materials and Methods (Figure 2G and H). Itraconazole induced accumulation of HUVECs in the G1 phase (88.3%) compared to the untreated control (82.1%), suggesting that itraconazole inhibits cell proliferation by arresting cells in the G1 phase.
Analysis of the mechanism of action of itraconazole in HUVECs. Next, we analyzed the mechanism of action of itraconazole through western blotting analysis of several cancer-related signaling pathways. In control HUVECs, we observed phosphorylation of p90RSK, S6, and p44/42 MAPK. AKT was not phosphorylated in untreated HUVECs. Interestingly, treatment with 1 μM itraconazole inhibited the phosphorylation of p44/42 MAPK and S6, whereas 100 μM itraconazole did not. Itraconazole did not inhibit the phosphorylation of p90RSK (Figure 3A).
Effects of itraconazole on the secretion of angiogenic cytokines from MKN-28 cells and CAFs. The Angiogenesis Antibody Array showed that MKN-28 cells secreted IL8, tissue inhibitors of metalloproteinase (TIMP) 1 and -2, chemokine (C-X-C motif) ligand 13, and uPA receptor (Figure 3B and C). VEGF was not detected in supernatants collected from MKN-28 cells. The secretion of these factors may be associated with the resistance of MKN-28 cells to bevacizumab. However, itraconazole did not influence the secretion of any of these factors in MKN-28 cells.
To evaluate the effects of itraconazole on the angiogenic factors secreted by CAFs in vitro using proteome arrays, we applied supernatants from CAFs to a Proteome Profiler Assay Human Angiogenesis Array (Figure 3D). Using this assay, we detected five angiogenic factors secreted from CAFs: IL1β, VEGF, TIMP1, MCP1, and PAI1. Moreover, of the five angiogenic factors detected in the array, only MCP1 secretion from CAFs was suppressed by itraconazole.
Effects of itraconazole and bevacizumab on the growth of tumors in mice. To determine the effects of these compounds in vivo, xenograft tumors from HT-29, MKN-28, and MKN-45 cells were established, and mice bearing these tumors were treated with bevacizumab, itraconazole, or a combination of both agents for 5 weeks. MKN-28-derived tumors responded poorly to bevacizumab treatment, whereas MKN-45-derived tumors showed significant decreases in tumor size following bevacizumab treatment (Figure 4). Tumors derived from HT-29 cells showed modest responses to bevacizumab treatment. From this result, we regarded MKN-45 cells as ‘bevacizumab responsive’, HT-29 cells as ‘modestly bevacizumab responsive’, and MKN-28 cells as ‘bevacizumab-resistant. Thus, we next investigated the anti-angiogenic and antitumor effects of itraconazole plus bevacizumab in bevacizumab-resistant MKN-28 cells. After 5 weeks of treatment, itraconazole alone did not elicit antitumor effects compared with the control group. Bevacizumab combined with itraconazole reduced the tumor size of MKN-28-derived tumors, which had been unaffected by bevacizumab alone. In HT-29-derived tumors, itraconazole alone did not elicit significant reductions in tumor volume after 5 weeks of treatment, with tumors having 88% of the volume of control tumors and 62% of the weight of control tumors.
Measurement of intratumoral microvessel formation revealed that bevacizumab alone did not reduce the formation of microvessels compared to the control group. However, bevacizumab combined with itraconazole caused a significant decrease in the formation of microvessels compared to that of the control group (p<0.0001).
Discussion
Itraconazole, an antifungal agent, is now widely and safely used for the treatment of diseases caused by fungi. It is also known for its antibleeding effects against pulmonary fungi in patients with rheumatoid disease who have hemosputum and hemorrhaging. Although this effect is thought to be mediated by the anti-angiogenic effects of itraconazole, the detailed mechanisms involved are not known. While possible mechanisms indeed involve the inhibition of angiogenic factors, our results also demonstrate that itraconazole has anti-angiogenic effects independent of VEGF. Our study suggests that the anti-angiogenic effects of itraconazole were due to its direct stimulation of apoptosis in endothelial cells, not due to inhibition of VEGF signaling. Similar to mTOR inhibitors, itraconazole blocked the phosphorylation of S6 and MAPK in HUVECs.
Another mechanism through which itraconazole exerted its anti-angiogenic effects was through the suppression of CAFs. To the best of our knowledge, no reports have previously demonstrated the influence of itraconazole on CAFs. CAFs play a critical role in both tumor progression and tumor angiogenesis. Kalluri et al. reported that myofibroblasts expressing α-SMA (CAFs) reportedly mediate CSI, essential for tumor growth (16). The CSI between cancer cells and CAFs influences bevacizumab resistance in tumors. Our results show that itraconazole suppressed both the growth of and the secretion of angiogenic factors from CAFs. Importantly, these mechanisms also affect the creation of new vascular networks. Therefore, we sought to determine whether treatment with itraconazole may help overcome any mechanisms of resistance associated with VEGF. Anti-angiogenic effects are thought to involve two mechanisms: one is the blockage of angiogenic factors, such as VEGF, and the other is inhibition of alternate pathways affecting angiogenesis. However, our results demonstrate the existence of a novel mechanism quite different from previous mechanisms, namely the direct inhibition of HUVEC and CAF proliferation.
Previous studies have shown that gastric cancer often exhibits resistance to bevacizumab (17). The MKN-28 cells used in this study were also resistant to bevacizumab. Our current results show that this resistance in MKN-28 cells (17) may be based on mechanisms involving other angiogenic factors, independently from VEGF. Indeed, treatment with bevacizumab did not affect the microvessel density in MKN-28-derived xenograft tumors in mice. However, even in bevacizumab-resistant gastric cancer cells, itraconazole reduced tumor volume and weight. Furthermore, the microvessel density of tumors treated with itraconazole significantly decreased compared to those in the control and bevacizumab-treated groups. Therefore, we conclude that this change in microvessel density resulted in the decreased tumor volume seen in itraconazole-treated mice.
Some investigators reported that itraconazole suppresses the proliferation of non-small cell lung cancer cells (18, 19). These effects have been tested in several clinical trials in non-small cell lung cancer and prostate cancer (12, 13). However, in our study, itraconazole did not inhibit the growth of gastrointestinal cancer cells. Nevertheless, resistance of these cells to bevacizumab has resulted in the contraindication of bevacizumab for use in gastric cancer (20), and acquisition of resistance to bevacizumab is one of the most debated topics in colorectal cancer therapy. Thus, the development of novel therapies to enhance bevacizumab sensitivity is an urgent issue. Our results demonstrate that itraconazole enhances the effects of bevacizumab, even in bevacizumab-resistant cancer cells. We believe that this synergistic effect of itraconazole may be an option for patients with bevacizumab-resistant colorectal cancer and may represent a novel therapy for gastric cancer.
Conclusion
In this study, we demonstrated that itraconazole had anti-angiogenic effects in addition to its known anti-fungal effects. This anti-angiogenic mechanism was mediated by inhibition of endothelial cell growth, a mechanism that is quite different from other well-known mechanisms, such as inhibition of VEGF or the CSI. Therefore, our study suggests that itraconazole may be useful for enhancing the anticancer effects of bevaciumab in the treatment of gastrointestinal cancer.
Footnotes
Conflicts of Interest
None.
- Received November 11, 2015.
- Revision received December 7, 2015.
- Accepted December 10, 2015.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Breaking the Crosstalk of the Cellular Tumorigenic Network in NSCLC by a Highly Effective Drug Combination
- Itraconazole targets cell cycle heterogeneity in colorectal cancer
- Molecular Effects of Stromal-Selective Targeting by uPAR-Retargeted Oncolytic Virus in Breast Cancer
- Severe Cardiotoxicity in a Patient with Colorectal Cancer Treated with Bevacizumab