


Abstract
Background/Aim: Prognosis for patients with head and neck squamous cell carcinoma (HNSCC) is poor in most cases and has not improved despite advances in therapy. Novel therapeutic approaches are mandatory in order to improve the situation. Everolimus, an inhibitor of mammalian target of rapamycin, as well as the multi-tyrosine kinase inhibitors sorafenib and sunitinib, has demonstrated a substantial therapeutic effect in various types of human cancer with moderate side-effects. Expression of vascular endothelial growth factor receptor (VEGFR) 1 and 2, and of the tumor-suppressor protein phosphatase and tensin homolog deleted on chromosome 10 (PTEN) were evaluated in chemonaïve human papillomavirus (HPV)-positive and -negative squamous cell carcinoma (SCC) and after exposure to everolimus, sorafenib or sunitinib. Materials and Methods: p16-positive CERV196 and p16-negative HNSCC 11A and 14C cells were incubated with different drug concentrations for 48-192 h. Expression of VEGFR1 and -2 as well as PTEN were determined by enzyme-linked immunosorbent assay and was compared to a chemonaïve control. Results: VEGFR1 and -2, as well as PTEN, were expressed in all three cell lines. Sunitinib, sorafenib and everolimus significantly reduced the expression of VEGFR1 and -2, especially in p16-positive CERV196 cells. Sunitinib appeared to be more effective in reducing VEGFR1 and -2 expression than sorafenib and everolimus. PTEN levels were remarkably lower in HPV-positive CERV196 cells. PTEN expression increased significantly under sunitinib and sorafenib in HNSCC 11A and CERV196 cells. Everolimus, on the other hand, led to a significant decrease of PTEN expression in these cell lines. Conclusion: The tested drugs displayed a remarkable anti-angiogenic effect by inhibition of VEGFR1 and -2 expression. Sunitinib and sorafenib were able to increase PTEN expression, which might induce apoptosis of cancer cells. HPV-positive CERV196 cells were characterized by an increased susceptibility to these small-molecule drugs. Further studies are imperative to scrutinize HPV status-dependent differences in drug response and possible implications for future treatment options.
Every year, 631,000 new cases of head and neck squamous cell carcinoma (HNSCC) are diagnosed, and approximately 352,000 patients die as a result of this tumor entity. This sixth most common cancer in the world is characterized by variable tumor aggressiveness and response to treatment (1). Prognosis and overall survival for the patient are poor and have not improved despite advancements in diagnostics, surgery, radiation and chemotherapy (2). The most important risk factors for HNSCC remain smoking, alcohol abuse and infection with human papillomavirus (HPV) (3-5). Recently, the incidence of HNSCC has decreased, which is attributed to the falling prevalence of tobacco abuse (6). On the contrary, the incidence of carcinomas of the tonsils and tongue base is rising in patients aged 20-44 years (7). These oropharyngeal carcinomas are indeed associated with HPV, and their rising incidence illustrates the importance of the viral etiology (8, 9).
Risk factors for HPV infection are promiscuity and unsafe sex. Thus HPV infections are associated with anogenital carcinomas, such as carcinomas of the uterine cervix, and oropharyngeal cancer (10). HPV-positive HNSCC must be differentiated from HNSCC associated with tobacco and alcohol abuse since they differ in molecular and clinical aspects (11-13). Viral oncogenes are able to suppress inflammation as one possible mechanism to generate tolerance by the immune system and avoid eradication (14). They degrade tumor-suppressor protein p53, which regulates the expression of angiogenic inhibitors and suppresses proangiogenic factors (15, 16). HPV-positivity in HNSCC is said to be a rather favorable factor because such carcinomas appear to be more sensitive to radiation and chemotherapy (17, 18).
Tumor cells, as well as tumor stromal cells, generate a tumor microenvironment that supports tumor growth and progression (19, 20). Angiogenic peptides, such as vascular endothelial growth factor (VEGF), regulate endothelial cell migration, proliferation and formation of capillaries in vivo. VEGF is associated with increased tumor growth and angiogenesis in HNSCC and displays anti-apoptotic potential (21, 22). HPV oncoprotein E7 stimulates expression of VEGF among other angiogenic factors (14). Vascular endothelial growth factor receptors (VEGFRs) are tyrosine kinase receptors that are activated by ligand–receptor interaction. VEGF and VEGFR expression are increased in various malignant tumors, such as breast cancer, melanoma and HNSCC (23, 24).
Tumor-suppressor proteins, such as p53 and phosphatase and tensin homolog deleted on chromosome 10 (PTEN), not only prevent the formation of tumor cells, but may also induce apoptosis of a tumor cell. They take part in cell cycle control and are often found to be disabled in human cancer cells. For instance, mutations of TP53, encoding for tumor-suppressor protein p53, occur in 45-70% of HNSCC cases (25). A.G. Knudson discovered the first tumor-suppressor protein, the retinoblastoma protein, in the 1970s. He described hereditary forms of retinoblastoma that occurred in young children and coined the ‘two-hit hypothesis’. According to this hypothesis, both alleles of a tumor-suppressor protein have to be mutated in order to lose their function (loss-of-function mutation) (26, 27). This is an important difference from oncogenes that become active by the mutation of a single allele (gain-of-function mutation). PTEN is a tumor-suppressor protein which negatively regulates the activity of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B signaling pathway, which can be induced by oncogenes such as Rat sarcoma protein. In this way, PTEN prevents suppression of apoptosis and inhibits cell growth and proliferation. It is said to be one of the most frequently altered signaling pathways in HNSCC (28). PTEN de-phosphorylates phosphatidylinositol-triphosphate (PIP3) into phosphatidylinositol-bisphosphate (PIP2) and prevents activation of protein kinase B. PTEN activity is known to be restricted in various types of human cancers (29). While PTEN mutations were identified in up to 16% of HNSCCs, loss of heterozygosity of PTEN appeared in up to 40% of HNSCCs, and loss of PTEN expression was found in about 29% of tongue cancer cases (30-32). Since increased phosphorylation of protein kinase B and reduced PTEN protein levels can be found in 70-80% of HNSCCs, a different mechanism of PTEN inhibition has to be assumed (33).
Given the poor prognosis of HNSCC, innovative pharmacological approaches are mandatory. The drugs tested in the present study have the advantage of oral application, moderate side-effects and a tumor-targeted mechanism of action.
The mammalian target of rapamycin (mTOR) belongs to the family of the PI3K-related kinases. By phosphorylation and activation of several other proteins, mTOR takes part in the regulation of cell growth, proliferation and motility, as well as in cell survival, gene transcription and protein synthesis. For instance, mTOR activates the protein kinase B signaling pathway (34). Consequently, mTOR antagonizes the effect of PTEN. mTOR alters DNA transcription and protein synthesis of, for instance, VEGF, and therefore induces angiogenesis (35, 36). The mTOR inhibitor everolimus is used as an immunosuppressant to prevent rejection of organ transplants (37) and as part of therapy regimens against several types of human cancer, including HNSCC (38-40). mTOR inhibitors showed antitumor activity in solid tumors that were resistant to VEGF/VEGFR-targeted therapy with bevacizumab (38). mTOR can affect various cellular signaling pathways found to be dysregulated in cancer (34, 39). This is reflected in the synergistic effects of mTOR inhibitors with other targeted-therapy drugs and radiochemotherapy (41-43). Lymph node metastases of HNSCC often exhibit an activated mTOR signaling pathway. Inhibition by everolimus reduces lymphangiogenesis at the primary tumor site and averts lymphatic dissemination (44).
Sorafenib and sunitinib are multi-tyrosine kinase inhibitors. Sorafenib targets VEGFR, platelet-derived growth factor receptor (PDGFR) and rat fibrosarcoma protein (Raf) kinases. It is a treatment option for hepatocellular, thyroid and renal cell carcinoma, lung cancer, melanoma and HNSCC (45-48). A synergistic effect of sorafenib in combination with radiation, chemotherapy or agents of targeted anticancer therapy has been observed (49, 50).
Sunitinib inhibits the activity of VEGFR, PDGFR, stem cell factor receptor, rearranged during transfection (RET) and colony stimulating factor. Established treatment regimens for gastrointestinal stromal tumors, renal cell carcinoma, pancreatic neuroendocrine tumors, breast, lung, thyroid and colorectal cancer include sunitinib (51, 52). Recently, sunitinib was evaluated for HNSCC therapy in clinical trials (53-55).
Everolimus, sorafenib and sunitinib have been evaluated for the treatment of HNSCC in several studies (42, 43, 53-58); however, data on HPV status-dependent differences are still missing. This study evaluated the protein expression of VEGFR1 and -2 and PTEN in p16-positive (CERV196) and p16-negative (HNSCC 11A and 14C) cell lines and the alteration of expression after incubation with everolimus, sorafenib, and sunitinib. To our knowledge, this is one of the first in vitro studies to investigate the effect of these small molecules on p16-positive and -negative squamous cell carcinoma (SCC).
Materials and Methods
Cell lines. The human cell lines HNSCC 11A and 14C were received from Dr T. E. Carey (University of Michigan, Ann Arbor, MI, USA) and originated from a SCC of the oropharynx and larynx. CERV196 cells were obtained from a p16-positive SCC of the uterine cervix (CLS, Eppelheim, Germany). Dulbecco's modified essential medium (DMEM) (Fisher Scientific and Co., Pittsburgh, PA, USA) supplemented with 10% fetal calf serum (FCS) and antibiotics (Life Technologies Inc., Gaithersburg, MD, USA) was used as medium for HNSCC 11A and 14C. CERV196 cells were kept in Eagle's minimum essential medium with 2 mM L-glutamine, 10% FCS and Earle's balanced salt solution with 1.5 g/l sodium bicarbonate, 0.1 mM amino acids and 1.0 mM sodium pyruvate. Cell cultures were carried out at 37°C in a fully humidified atmosphere with 5% CO2. Everolimus (Novartis, Basel, Switzerland), sorafenib (Bayer, Leverkusen, Germany) and sunitinib (Pfizer, New York, NY, USA) were dissolved in dimethylsulfoxide (DMSO) at the time of use. The cells were exposed to drug concentrations ranging from 1.0-25.0 μmol/ml for 2 to 8 days. The drug concentrations were chosen after performing the alamarBlue (AbD Serotec, Oxford, UK) cell proliferation assay, which measured the proliferation of HNSCC tumor cell lines quantitatively and indicated the relative cytotoxicity of the studied drugs.
Enzyme-linked immunosorbent assay (ELISA) for total VEGFR1 and -2 and PTEN. After incubation, treated cells were rinsed with phosphate-buffered saline (PBS), and 350 μl of lysis buffer were added to each well. The lysed cells were stirred up with a vortex for 30 min at 2-8°C and microcentrifuged for 5 min at 2,000 × g. The supernatant was pipetted into a clean tube.
Protein concentrations were determined with the ELISA technique. We used DuoSet IC Human Total VEGFR 1 (DYC4347), DuoSet IC Human Total VEGFR 2 (DYC1780) and DuoSet IC Human Total PTEN (DYC847) kits (R&D Systems, Wiesbaden, Germany).
The sandwich ELISA system used a solid-phase capture antibody specific for human VEGFR1, VEGFR2 or PTEN and a specific detection antibody using a standard streptavidin-horseradish peroxidase (HRP) format. The capture antibody was diluted to the working concentration (0.4 μg/ml for VEGFR1; 0.2 μg/ml for VEGFR2; 0.05 μg/ml for PTEN). Then 100 μl of the capture antibody were added to each well and plates were incubated overnight. The contents of each well were then aspirated, and the wells were washed three times with 400 μl of Tween buffer. Then 300 μl of block buffer were added to each well and plates further incubated for 1-2 h followed by another Tween buffer wash as previously described. The detection antibody was diluted to its working concentration (0.4 μg/ml for VEGFR1; 0.2 μg/ml for VEGFR2; 0.05 μg/ml for PTEN). Then 100 μl of the detection antibody were added to each well and plates were incubated for 2 h at room temperature. The wells were washed again as previously described, and 100 μl of streptavidin-HRP (diluted according to instructions) were added to each well followed by 20 min of incubation at room temperature. The wells were washed again. Afterwards, 100 μl of substrate solution were added to each well for 20 min followed by 50 μl of stop solution. According to the manufacturer's directions, each ELISA was performed with 100 μl of supernatant. All analyses and calibrations were carried out three times. The calibrations on each microtiter plate included recombinant human VEGFR1, VEGFR2 and PTEN standards that were provided in the kits. Optical density was measured with a microplate reader at a wavelength of 450 nm. Wavelength correction was set to 540 nm, and concentrations are reported as pg/ml. The range of detection was 312-20,000 pg/ml for VEGFR1, 62.5-4,000 pg/ml for VEGFR2, and 312-20,000 pg/ml for PTEN. The interassay coefficient of variation reported by the manufacturer was below 10%.
Measuring total protein. Total protein was measured with DC Protein Assay (BioRad, Hercules, CA, USA). Cells were incubated, lysed and centrifuged as previously described. Dilutions of protein standard were prepared according to the manufacturer's instructions. Measurement was performed on 100 μl of protein standard or cell supernatant with a spectrophotometer set to 750 nm, and concentrations are reported as μg/ml.
Statistical analysis. Statistical analysis was carried out in cooperation with PD Dr. C. Weiss, Institute of Biomathematics, Faculty of Medicine, Mannheim, Germany. A p-value ≤0.05 was considered statistically significant. The two-coefficient variance test (SAS Statistics, Cary, NC, USA) and Dunnett's test were used.
Results
Total protein assay. Total protein assay was performed in order to differentiate a reduction of the expression of PTEN and VEGFR1 and -2 from a cytotoxic effect of the applied drugs. The total protein level in the cell lysate was compared to the expression levels of the target values. We detected only minor fluctuations of the protein quotient (expression of the target value/total protein level). An increase of the quotient would indicate an increase of apoptosis under incubation with the drugs. However, an increase of this quotient was not detected irrespective of HPV status, target protein, applied drug or drug concentration. Consequently, alterations of protein expressions found cannot be explained by cytotoxicity of the applied drugs (data not shown).
ELISA for VEGFR1 and -2 expression in HNSCC 14C, 11A and CERV196 cells. Similar levels of VEGFR1 were expressed in all cell lines independent of the HPV status. VEGFR1 showed a trend towards decreased expression in HNSCC 11A and 14C cells after incubation with everolimus, sunitinib and sorafenib. However, no statistical significance in differences was detected. On the contrary, in p16-positive CERV196, significant suppression of VEGFR1 levels was found with everolimus, sunitinib and sorafenib. The effect of sunitinib and sorafenib occurred in an incubation-time dependent manner. Everolimus suppressed VEGFR1 levels by 8.7% (p-values varied between 0.001 and 0.007). Incubation with sunitinib led to an average VEGFR1 suppression in CERV196 of 31.1% (p-values <0.001-0.012). The p-values for incubation with sorafenib ranged between 0.003 and 0.013, and the average suppression of VEGFR1 in CERV196 was 18.0%. Sunitinib was more effective at suppression of VEGFR1 expression compared to sorafenib, but this effect was not statistically significant. In HNSCC 11A and 14C cells, increasing the drug concentration had no effect on the alteration of VEGFR1 expression. In CERV196 cells, a trend towards stronger VEGFR1 suppression was observed after incubation with higher drug concentrations, but statistical significance was not reached. For simplification, only the data for treatment at 25 μmol/ml are shown in Table I and Figure 1.
VEGFR2 expression was considerably lower compared to VEGFR1 independent of the cell line. In HNSCC 11A cells, a significant decrease of VEGFR2 was detected with everolimus after 3 days of incubation (p=0.014). On average, everolimus suppressed VEGFR2 levels by 13.1%. For sunitinib and sorafenib, a trend towards reduced expression of VEGFR2 was observed but statistical significance was not reached. Suppression of VEGFR2 expression appeared to be incubation time-dependent. Similar results were observed for HNSCC 14C cells. A significant decrease of VEGFR2 expression was detected after 2 and 5 days of incubation with sunitinib (p=0.001 and <0.0001, respectively). Again, VEGFR2 suppression appeared to be incubation time-dependent. In CERV196 cells, everolimus significantly reduced VEGFR2 expression independent of incubation time (p=0.006-0.036). Average VEGFR2 suppression by everolimus was 15.8%. Sunitinib significantly suppressed VEGFR2 after 2 days of incubation (p=0.049). For sorafenib, a significant decrease of VEGFR2 was detected after 2, 3 and 8 days of incubation (p=0.005, 0.01 and 0.047, respectively). Again, sunitinib appeared to be slightly more effective at VEGFR2 suppression, but without statistical significance. Increasing the drug concentrations had no additional significant effect. Only the data for treatment at 25 μmol/ml are shown in Table II and Figure 2.
ELISA of PTEN expression in HNSCC 14C, 11A and CERV196 cells. The tumor-suppressor protein PTEN was expressed in all three cell lines. A total loss of PTEN expression was not found in either HPV-positive or HPV-negative SCC. However, CERV196 cells were found to have lower PTEN levels (about 30% of the level measured in HNSCC 11A and 14C cells) compared to HPV-negative SCC. In HNSCC 11A cells, everolimus significantly reduced PTEN expression after 2, 3 and 5 days of incubation (p=0.015, 0.018 and 0.040, respectively). Both sunitinib and sorafenib increased PTEN expression in HNSCC 11A cells. Surprisingly, this effect was smaller after 8 days of incubation; even reduction in the level of PTEN expression was detected. Sunitinib significantly increased PTEN expression after 3, 5 and 8 days of incubation (p=0.044, 0.044 and 0.011, respectively). The average increase of expression was 5.1%. For sorafenib, we detected a significant increase after 2, 3 and 5 days of incubation (p=0.014, 0.007 and 0.019, respectively). After 8 days of incubation with sorafenib, PTEN levels decreased significantly by 2.6% (p=0.020). In HNSCC 14C cells, everolimus showed a trend towards reducing PTEN expression in an incubation time-dependent manner, but no significant alteration was detected. Sunitinib increased PTEN expression (by 4.1% on average) compared to the negative control. However, statistical significance was detected after 2 days of incubation only (p=0.022). Similar to HNSCC 11A cells, this effect could not be detected after 8 days of incubation. Sorafenib reduced PTEN expression in HNSCC 14C cells but this effect was not significant. As in HNSCC 11A and 14C cells, everolimus reduced PTEN expression in CERV196 cells. Statistical significance was reached after 2, 3 and 5 days of incubation (p=0.027, 0.048 and 0.003, respectively). Sunitinib increased PTEN expression after 2 days and significantly increased expression after 3 days (p=0.027). After 5 and 8 days of incubation, we detected reduction of PTEN level (p-value=0.001). Following incubation with sorafenib, PTEN levels significantly increased in CERV196 cells after 2, 3 and 5 days (p=0.014, 0.005 and 0.023, respectively). Again, this effect was not detected after 8 days of incubation. Here we found a significant decrease of PTEN level (p=0.006). Higher drug concentrations were significantly more effective at altering PTEN expression, especially in CERV196 and HNSCC 11A cells. Data are shown in Table III and Figure 3.
Discussion
This study sought to evaluate the expression of VEGFR1 and -2 and PTEN in the cell lines CERV196 (p16-positive), HNSCC 11A and 14C (p16-negative) and the impact of the mTOR inhibitor everolimus and the two multi-tyrosine kinase inhibitors sorafenib and sunitinib on VEGFR1 and -2 and PTEN expression.
The formation of new blood vessels is one condition for tumor progression, further invasion and the formation of lymph node and distant metastases. It is reasonable to target proangiogenic factors in anticancer therapy since there is a correlation between microvessel density in a tumor and its recurrence (59, 60). The suppression of VEGFR1 and -2 could, therefore, help reduce blood supply for a tumor. The structural and functional abnormalities of tumor vessels lead to a chaotic blood flow and can disturb oxygen supply for the tumor. A reduction of this chaotic blood flow by antiangiogenic therapy may increase tumor radiosensitivity during combined radiochemotherapy (61, 62).
We were able to demonstrate that the inhibitors of VEGFR tyrosine kinase, sunitinib and sorafenib, as well as the mTOR inhibitor everolimus reduce expression of VEGFR1 and -2. This effect was incubation time-dependent, and sunitinib was slightly but not significantly more effective compared to sorafenib and everolimus. In HNSCC 11A and 14C cells, we detected reduction of VEGFR1 after incubation with sunitinib, sorafenib and everolimus, but statistical significance was not reached. Similar findings can be stated for the expression of VEGFR2 in HNSCC 11A and 14C cells. With the exception of everolimus in HNSCC 11A after 3 days of incubation and of sunitinib in HNSCC 14C after 2 and 5 days of incubation, the decrease of VEGFR2 expression was not significant but appeared to be incubation time-dependent. Surprisingly, we detected a statistically significant reduction of VEGFR1 in p16-positive CERV196 cells only. Although everolimus is not a direct inhibitor of VEGFR, we can state there was a significant decrease of VEGFR1 and -2 expression in CERV196 cells independently of the incubation time when compared to the negative control. Lane et al. reported that everolimus indeed has anti-angiogenic properties and reduces VEGF release from tumor cells. These effects are similar but also distinct from those of VEGFR tyrosine kinase inhibitors (63). During a targeted-therapy, the selection of resistant cancer cells is a possible mechanism for therapy failure. This was reported for anti-epidermal growth factor receptor therapy with cetuximab or antiangiogenic VEGFR-targeted drugs (64, 65). Both sorafenib and sunitinib are able to overcome such drug resistance. This can be explained by their wider inhibition of several tyrosine kinases downstream of the receptor molecule (65, 66).
Enzyme-linked immunosorbent assay of Vascular endothelial growth factor receptor 1 (VEGFR1) expression (pg/ml) in head and neck squamous cell carcinoma (HNSCC) 11A, 14C and CERV196 cells after incubation with everolimus, sunitinib or sorafenib (at 25 μmol/ml) compared to the negative control (statistical significance is indicated in bold).

Vascular endothelial growth factor receptor 1 (VEGFR1) expression in head and neck squamous cell carcinoma (HNSCC) 11A, 14C and CERV196 cell lines after incubation with everolimus, sunitinib or sorafenib compared to the negative control. Data are mean values±standard deviation.
Enzyme-linked immunosorbent assay of Vascular endothelial growth factor receptor 2 (VEGFR2) expression (pg/ml) in head and neck squamous cell carcinoma (HNSCC) 11A, 14C and CERV196 cells after incubation with everolimus, sunitinib or sorafenib (at 25 μmol/ml) compared to the negative control (statistical significance is indicated in bold).
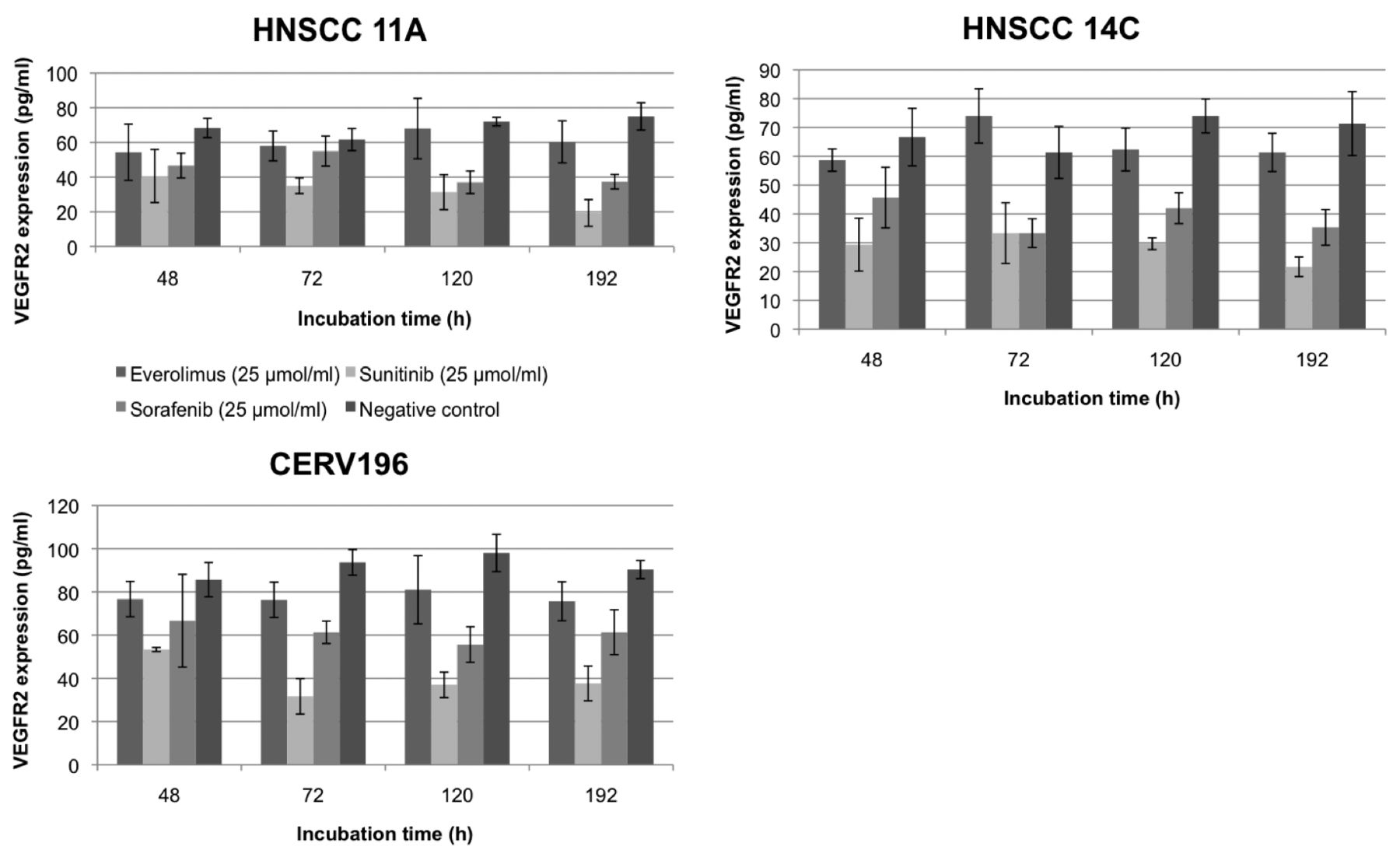
Vascular endothelial growth factor receptor 2 (VEGFR2) expression in head and neck squamous cell carcinoma (HNSCC) 11A, 14C and CERV196 cells after incubation with everolimus, sunitinib or sorafenib compared to the negative control. Data are mean values±standard deviation.
Enzyme-linked immunosorbent assay of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) expression (pg/ml) in head and neck squamous cell carcinoma (HNSCC) 11A, 14C and CERV196 cells after incubation with everolimus, sunitinib or sorafenib (at 25μmol/ml) compared to the negative control (statistical significance is indicated in bold).

Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) expression in head and neck squamous cell carcinoma (HNSCC) 11A, 14C and CERV196 cells after incubation with everolimus, sunitinib or sorafenib compared to the negative control. Data are mean values±standard deviation.
The expression levels of VEGFR2 were considerably lower than VEGFR1 levels in all tested cell lines. Similar results have been reported on the expression of PDGFRα and PDGFRβ in these cell lines (67). More importantly, the results on PDGFRα and -β confirm our finding that CERV196 cells have a higher sensitivity to a small molecule-induced decrease of proangiogenic factors, which could be evidence that p16-positive HNSCCs have a higher sensitivity to targeted antiangiogenic therapy compared to p16-negative carcinomas (67).
As previously mentioned, mutations of PTEN can be detected in up to 16% of HNSCCs, loss of heterozygosity occurs in about 40% and PTEN loss can be found in 29% of HNSCCs (30-32). None of our cell lines showed loss of PTEN expression. Thus, PTEN can be a possible target for therapy in HNSCC. Interestingly, the p16-positive cell line CERV196 exhibited a considerably lower PTEN level (30%) when compared to the p16-negative cell lines HNSCC 11A and 14C. Both sunitinib and sorafenib significantly increased PTEN expression in HNSCC 11A and CERV196 cells. A minor increase of PTEN expression was detected in HNSCC 14C cells after incubation with sunitinib, but this effect was not significant. This increase of PTEN expression following incubation with sunitinib confirms the results of Abouantoun et al. in medulloblastoma cells (68). Ruan and colleagues reported that PTEN enhances the sensitivity of hepatocellular carcinoma cells to sorafenib (69). Combined with our results, this could imply a possible mechanism of self-enhancement of the effect of sorafenib. Interestingly, the drug effect for increased PTEN expression diminished or even disappeared in all cell lines after 5 or 8 days of incubation. Yung and colleagues reported similar findings in gastrointestinal stromal tumors after long-term exposure to sunitinib by epigenetic PTEN gene silencing (70). This is a possible mechanism of tumor drug resistance to multi-tyrosine kinase inhibitors. Incubation with everolimus significantly decreased the PTEN level in HNSCC 11A and CERV196 cells independently of the incubation time. HNSCC 14C cells showed no alteration of PTEN expression following incubation with everolimus. Seront et al. demonstrated that PTEN deficiency is associated with a reduced sensitivity to mTOR inhibitors (71). Similar results were published by Yang et al. (72). As previously described, mTOR activates the AKT signaling pathway and antagonizes the effect of PTEN that way (34). Our findings that PTEN expression decreased after incubation with everolimus in HNSCC 11A and CERV196 cells could be a possible evasive mechanism of the cancer cells for this drug.
Recent molecular analysis of common forms of cancer revealed that these diseases include a wide range of possible genetic alterations. The hope for a personalized anticancer treatment may lead to a paradigm shift in drug therapy from standard therapy regimens to an individualized therapy plan. The key problems with this shift are the identification and selection of oncogenic driver genes among numerous genetic alterations and the cellular heterogeneity of individual tumors, which may lead to the selection of resistant cancer cells during therapy (73). Consequently, personalized cancer treatment would require for continuous adaption of the therapy according to the tumor biology (74). Various phase II studies that investigated sunitinib and sorafenib as a single-drug application in HNSCC found little or no antitumor effect of these drugs. Therefore, sunitinib and sorafenib should not be used in monotherapy for HNSCC (53-57). HNSCC cells may develop various evasive mechanisms as discussed earlier that could lead to therapy failure. On the other hand, these novel agents can be part of a combination therapy. Sunitinib combined with cetuximab and radiation therapy significantly reduced cell proliferation (42). Everolimus was successfully used in combination with cisplatin and docetaxel (43, 58).
A personalized, targeted anticancer therapy in the future would require pre-therapeutical screening of the tumor for possible target proteins. A combination therapy with multiple targets would reduce the risk of therapy failure and the selection of resistant cancer cells. A possible evasive mechanism of the tumor cells would require control of drug efficacy during therapy.
As far as we are aware, this is one of the first studies to investigate the impact of everolimus, sunitinib and sorafenib on HPV-positive and -negative SCC in vitro. The study displayed the significant anti-angiogenic effects of these drugs, showed their potential to temporarily increase PTEN expression and confirmed an increased susceptibility of HPV-positive SCC to targeted-therapy. Further studies are imperative to assess the applicability of everolimus, sunitinib and sorafenib to HNSCC therapy and should distinguish the HPV status of tumor cells.
Acknowledgements
The Authors would like to thank Petra Prohaska for her outstanding technical assistance and PD Dr. C. Weiss for her distinguished advice in statistical analysis.
- Received October 25, 2014.
- Revision received November 7, 2014.
- Accepted November 13, 2014.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}