


Abstract
Glomus tumor is a rare perivascular neoplasm that usually occurs in the distal extremities of young adults. Recent molecular studies have identified microRNA 143–NOTCH fusions or NOTCH1-3 rearrangements in benign and malignant glomus tumors. Herein, we describe the cytogenetic and molecular cytogenetic findings of a glomus tumor arising in the left wrist of a 45-year-old man. Physical examination showed a 1.3-cm, blue-red, tender nodule. Magnetic resonance imaging demonstrated a subcutaneous, well-circumscribed mass with low signal intensity on T1-weighted sequences and high signal intensity on T2-weighted sequences. Contrast-enhanced fat-suppressed T1-weighted sequences showed a homogeneous, strong enhancement. A marginal excision was performed and histopathological examination confirmed the diagnosis of a glomus tumor. Cytogenetic and spectral karyotypic analyses showed a novel rearrangement involving chromosome bands 1p13 and 5q32. There has been no evidence of local recurrence four months after surgery. To the best of our knowledge, this is the first case of sporadic glomus tumor with t(1;5).
Glomus tumor is a generally benign pericytic tumor comprising fewer than 2% of soft tissue tumors (1). It typically presents as a small, blue-red, painful nodule in the distal extremities, particularly the subungual region of the finger. Multiple lesions may be seen in 10% of patients (1). Conventional glomus tumor is adequately managed by simple excision.
Histologically, the lesion is circumscribed and consists of uniform, round, or slightly polygonal epithelioid-like cells with sharp cellular borders and pale to bright eosinophilic appearance. In 2001, Folpe et al. defined the following histological criteria for malignancy in glomus tumor: large size (>2.0 cm) and deep location or moderate to high nuclear grade and increased mitotic rate (>5 per 50 high-power fields) or the presence of atypical mitotic figures (2). Immunohistochemically, the tumor cells are positive for vimentin and smooth muscle actin but negative for desmin, keratins and S-100 protein (1). Collagen IV-positive basement membranes are typically detectable around the tumor cells.
Only five cases of neurofibromatosis type 1 (NF1)-associated glomus tumor have been cytogenetically characterized in the literature (3). In the current study, we describe the first case, as far as we are aware of, of sporadic glomus tumor with clonal chromosomal aberrations occurring in the left wrist of a middle-aged man.
Case Report
A 45-year-old man presented with a 10-year history of increasing pain in the dorsal-ulnar aspect of the left wrist. Approximately 1.5 years before presentation, the patient underwent an attempted excisional biopsy of the lesion. The histological diagnosis at that time was glomus tumor. Subsequently, the patient had no improvement of symptoms. In fact, the nodule increased in size and became more painful to the touch in a few months after biopsy.
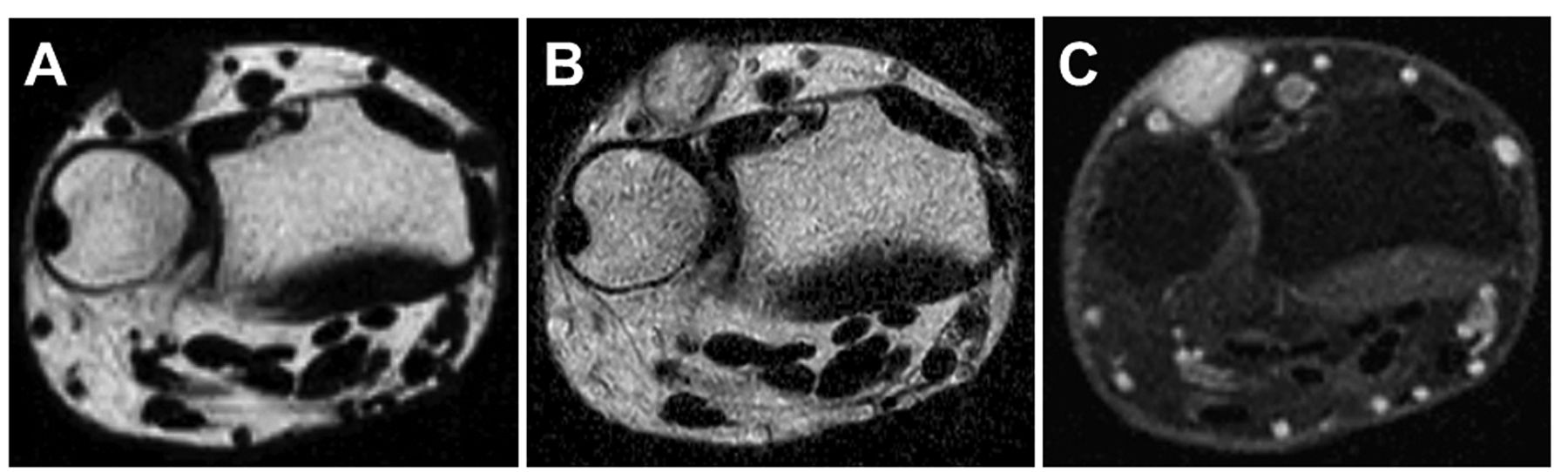
Physical examination revealed a 1.3-cm, blue-red, tender nodule. Magnetic resonance imaging (MRI) demonstrated a subcutaneous, well-circumscribed mass with low signal intensity on T1-weighted sequences (Figure 1A) and high signal intensity on T2-weighted sequences (Figure 1B). Contrast-enhanced fat-suppressed T1-weighted sequences showed homogeneous, strong enhancement (Figure 1C).
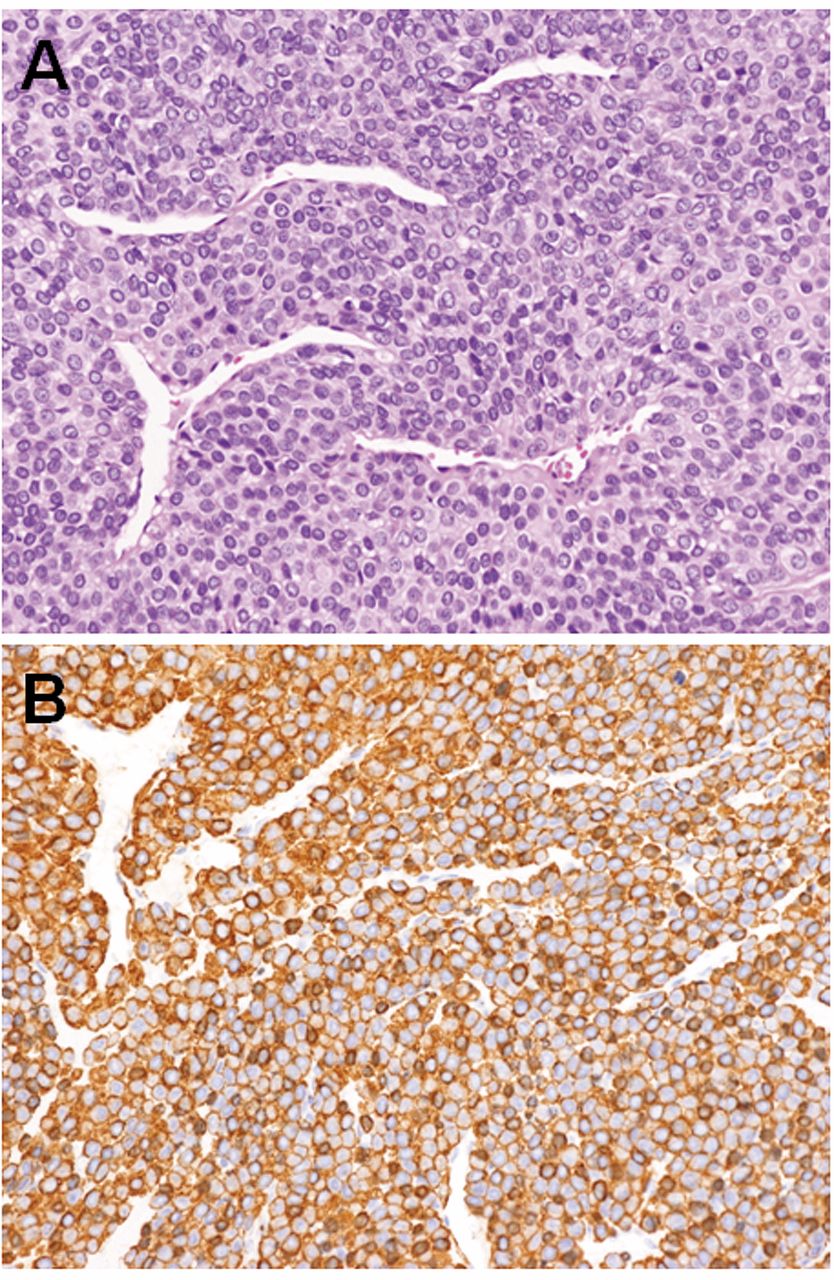
A marginal excision of the tumor was carried-out. Microscopically, the tumor was composed of a proliferation of uniform cuboidal cells arranged in nests with intervening dilated or slit-like blood vessels (Figure 2A). Neither cellular atypia nor atypical mitotic figures were observed. Immunohistochemically, the tumor cells were diffusely positive for smooth muscle actin (Figure 2B) but negative for cytokeratin. The tumor cells were surrounded by type IV collagen. Based on these findings, the tumor was diagnosed as a glomus tumor.

Axial magnetic resonance images of glomus tumor involving the dorsal-ulnar aspect of the left wrist. The subcutaneous mass had low signal intensity on T1-weighted image (A) and high signal intensity on T2-weighted image (B). Contrast-enhanced fat-suppressed T1-weighted image (C) revealed homogeneous, strong enhancement of the mass.
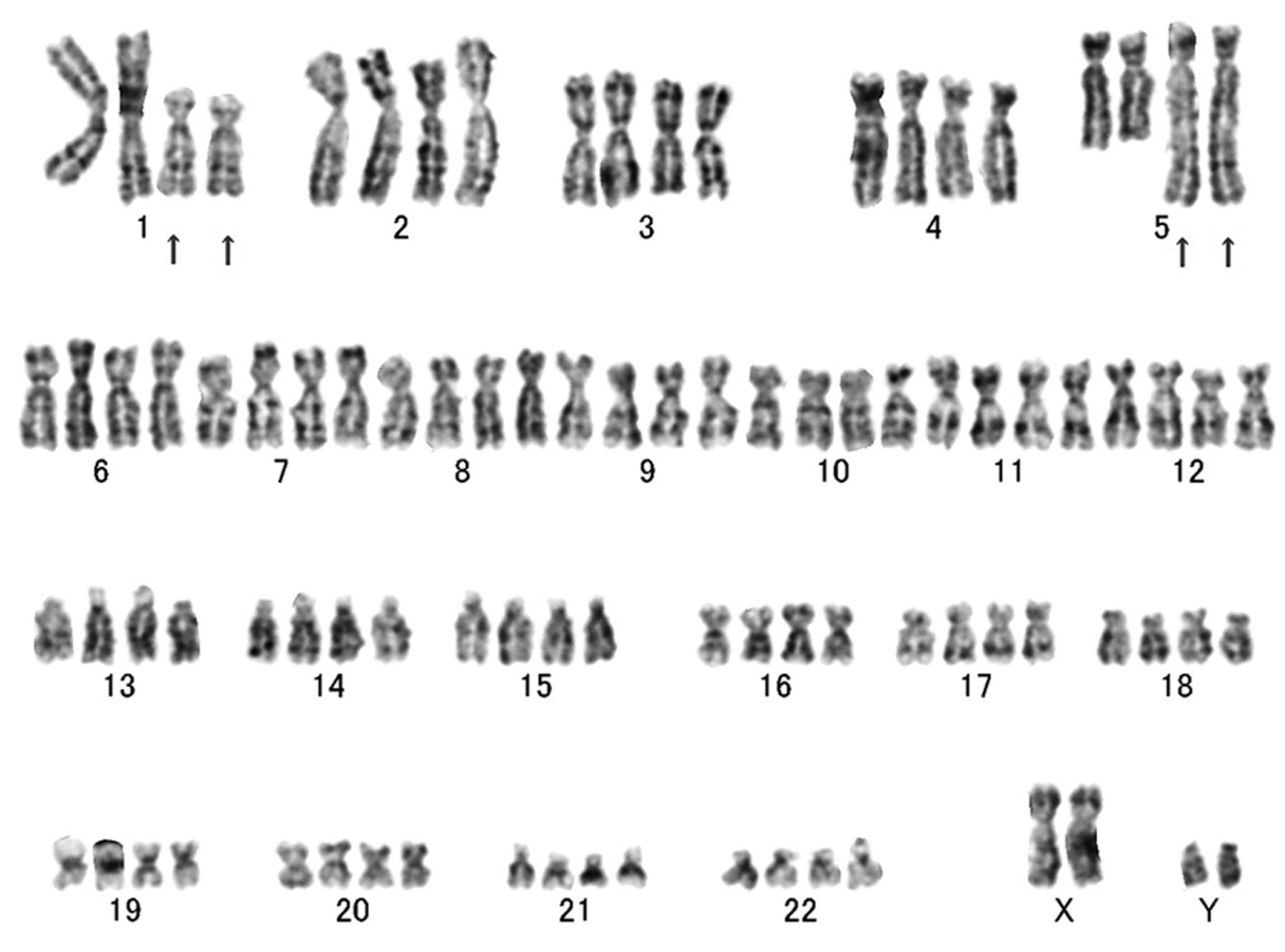
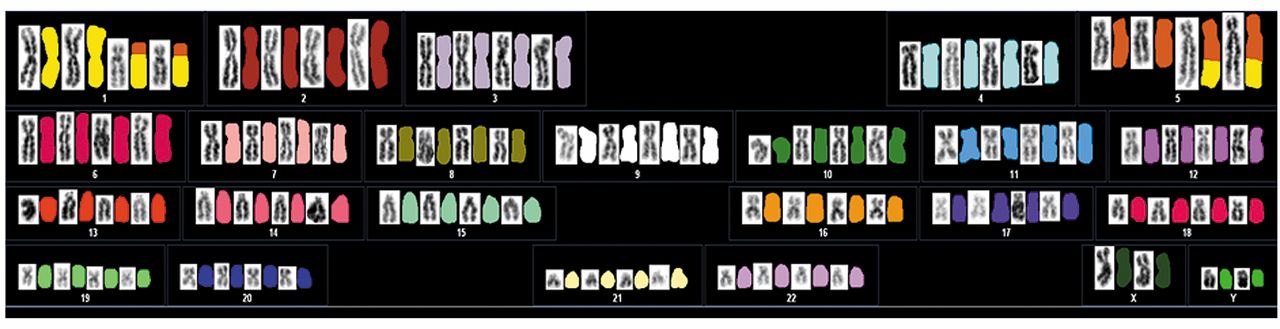
Conventional cytogenetic analysis revealed the following karyotype: 47,XY,add(1)(p13),add(5)(q32),+mar1[2]/92, idemx2, -mar1x2[8]/46,XY[10] (Figure 3). Spectral karyotyping (SKY) demonstrated the presence of a t(1;5)(p13;q32) translocation (Figure 4).
The postoperative course was uneventful and the pain was relieved immediately after surgery. At the 4-month follow-up, the patient was asymptomatic and there was no evidence of local recurrence.
Discussion
Through RNA sequencing, a novel microRNA 143 (MIR143)–NOTCH fusion has been detected in more than half of all glomus tumors, regardless of malignancy and anatomical location (4). MIR143 is located on chromosome 5q32 and acts a tumor-suppressor gene. The MIR143/145 bicistronic gene has been demonstrated to contribute to smooth muscle cell differentiation and proliferation (5). NOTCH2 is located on chromosome 1p11-13. NOTCH2 gene rearrangements have been identified in 52% of glomus tumors (4) and a small subset of breast cancer cell lines (6). More importantly, this gene fusion has never been recorded in mesenchymal neoplasms.
In the current study, we identified a novel t(1;5)(p13;q32) translocation using G-banding and SKY analyses. Our finding strongly supports the molecular data of Mosquera et al. (4). On the other hand, mitotic recombination of chromosome arm 17q has been identified as somatic inactivation mechanism in a subset of NF1-associated glomus tumors (3). A familial variant of glomus tumor (glomuvenous malformation) has been linked to chromosome 1p21-22 and involves truncating mutations in the glomulin gene (7).

Histological and immunohistochemical findings of glomus tumor. A: The tumor was composed of uniform cuboidal cells with round to oval nuclei (hematoxylin and eosin staining, original magnification ×100). B: Immunohistochemically, the tumor cells were diffusely positive for smooth muscle actin (original magnification ×100).

A representative G-band karyotyping of glomus tumor displaying rearrangements of 1p13 and 5q32. Arrows indicate the structural chromosomal aberration.

Spectral karyotyping of glomus tumor illustrating the 1;5 translocation. Classified image is displayed alongside the reverse DAPI image.
The main differential diagnosis includes angioleiomyoma. Angioleiomyoma typically presents as a small, slow-growing, firm, painful nodule in the extremities, particularly the lower leg. It has a peak incidence in the fourth to sixth decade of life, with a female predominance. On MRI, the tumor is usually isointense or slightly hyperintense relative to skeletal muscle on T1-weighted sequences and has a heterogeneous appearance on T2-weighted sequences. With administration of intravenous contrast material, strong enhancement with a heterogeneous or homogenous pattern is seen (8). To date, only six cases of angioleiomyoma have been cytogenetically characterized in the literature (9-13). Angioleiomyoma is usually diploid with no consistent chromosomal aberration. Previously, we applied comparative genomic hybridization to angioleiomyomas to screen for gains and losses of DNA sequences (14). The most frequent genetic change was a loss of 22q11.2.
In summary, we described the first case of glomus tumor with a novel t(1;5) translocation. Further studies are needed to elucidate the significance of this chromosomal alteration in the pathogenesis of glomus tumor.
Acknowledgements
This study was supported in part by the Foundation for the Promotion of Medical Science and JSPS KAKENHI Grant Number 25462355.
- Received July 14, 2015.
- Revision received August 21, 2015.
- Accepted August 26, 2015.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.