


Abstract
Cannabinoids possess a number of characteristics that make them putative anticancer drugs, and their value as such is currently being explored in a number of clinical studies. To further understand the roles that cannabinoids may have, we performed gene expression profiling in glioma cell lines cultured with cannabidiol (CBD) and/or Δ9-tetrahydrocannabinol (THC), and pursued targets identified by this screening. Results showed that a large number of genes belonging to the heat shock protein (HSP) super-family were up-regulated following treatment, specifically with CBD. Increases were observed both at the gene and protein levels and arose as a consequence of increased generation of ROS by CBD, and correlated with an increase in a number of HSP client proteins. Furthermore, increases impeded the cytotoxic effect of CBD; an effect that was improved by co-culture with pharmacalogical inhibitors of HSPs. Similarly, culturing glioma cells with CBD and HSP inhibitors increased radiosensitivity when compared to CBD-alone. Taken together, these data indicate that the cytotoxic effects of CBD can be diminished by HSPs that indirectly rise as a result of CBD use, and that the inclusion of HSP inhibitors in CBD treatment regimens can enhance the overall effect.
The number of clinical trials testing the activity of compounds known as cannabinoids has increased over the past two years. These trials have emerged as a direct consequence of the large amount of pre-clinical data generated recently in in vitro and in vivo studies that have highlighted the anticancer effects of cannabinoids (1). The cannabinoids are a group of inter-related chemicals extracted from the cannabis plant, out of which there exist approximately 80 in number. The two lead clinical compounds from this group are cannabidiol (CBD) and Δ9-tetrahydrocannabinol (THC) (2). Although these two compounds are structurally very similar, they possess distinct mechanisms of action. However, their ultimate effect tends to be induction of cell death or an arrest of cell growth. THC generally requires binding to a cognate cannabinoid receptor to elicit responses, whilst CBD appears to work independently of said receptors (3). This may explain why the dominant route into apoptosis varies in CBD and THC; with inhibition of AKT signaling and induction of autophagy being the cause of apoptosis for THC (4), whilst the generation of reactive oxygen species (ROS) and the ensuing involvement of mitochondria underlies the way that CBD can stimulate the intrinsic apoptosis pathway (5).
A large number of these current trials are performed in patients with glioma. This is because they respond well to cannabinoids - potentially because the cells tend to express the cannabinoid receptors that are generally thought to be required for activity. However, because the development of cannabinoids as putative anticancer drugs is still at an early stage, it remains important to determine the best treatment regimen. One way to achieve this is to better understand the mechanisms through which the agents work. Cannabinoid action is known to be multi-modal in nature because each compound is capable of modulating central signalling cascades such as ERK, PI3K and JNK (6), and in addition to their anti-proliferative and cytotoxic effects, they also possess anti-angiogenic, anti-migratory and anti-inflammatory characteristics. This diversity in action can be a great benefit to a compound and can be exploited in combination strategies by specifically identifying other treatments with mechanisms of action that are not impeded by or impede cannabinoid activity, which would ultimately lead to non-antagonistic interactions. Indeed, we have already demonstrated how THC can synergize with common leukaemia drugs such as cytarabine and vincristine, leading to enhanced anticancer responses (7), yet how combining it with cisplatin did not achieve the same response (8). This suggests that drugs that ultimately function through the cell cycle such as cytarabine (G1) and vincristine (G2/M) may be better partners with THC than drugs that directly elicit a “kill” signal such as cisplatin.
The heat shock proteins (HSPs) are a ubiquitous collection of proteins that are constitutively expressed and perform many essential functions. Their chaperone profile allows them to be primitive transporters that shuttle de novo synthesised proteins between sub-cellular organelles (9), but they also have a role in protein assembly, secretion and degradation as well as in the regulation of various transcription factors. One of their most recognised roles, however, is their importance in cell survival following exposure to various cellular stressors, such as temperature, pH and inflammation (10). These disrupt the homeostasis of a cell and can be induced by certain chemicals, but more often are caused by biological conditions such as hypoxia, hyperthermia and oxidative stress. The stressors cause fundamental alterations to the tertiary structures of proteins, and this ultimately disturbs cellular coherence. There are a number of HSPs that counteract this cellular disturbance and correct the misfolded/denatured proteins. They differ in the methods through which they achieve this, which defines the class within which they fall, but their ultimate outcome is targeted towards cellular protection, repair and survival. In this role, HSPs can either act as molecular chaperones, whereby they directly mend misfolded proteins and restore homeostasis (11), or they are able to sequester faulty or irreparable proteins by targeting them for degradation through ubiquitination, and thus prevent their interactions with others. Their client proteins are varied but typically promote survival; therefore, as a consequence of the protection of these proteins, HSPs can also inadvertently maintain cancer survival. The increase in HSPs in tumour cells may thus be an undesirable effect, and indeed can be used as a prognostic marker (12). This also explains why therapeutic approaches have been developed that attempt to minimize the effects of HSPs in cancer cells (12).
As part of our ongoing program of research into the potential therapeutic benefits of cannabinoids, we have endeavoured to further understand the distinctions in the mechanisms of actions of the two clinical lead cannabinoids, CBD and THC. To do this, we have performed gene expression screens using glioma cell lines to discern the genetic targets of CBD and/or THC. From these studies, we identified that HSPs are implicated in the actions of CBD, but not THC, and have subsequently attempted to understand the reason for this. Using our knowledge of HSPs and the role they play in protection and survival, we were then able to select the appropriate combination partners for CBD that would enhance its effects. We tested these predications through the use of combination models and molecular profiling and indeed successfully improved the efficacy of the cannabinoid. Overall, these studies will hopefully guide the development and use of CBD, which is especially pertinent as this compound has recently entered phase 1b/2 clinical trials.
Materials and Methods
Cell culture and cannabinoids. Two human glioma cell lines T98G (glioblastoma multiforme) and U87MG (glioblastoma astrocytoma) were purchased from the European Collection of Cell Cultures (Salisbury, UK) and were grown in DMEM (Sigma-Aldrich Company Ltd., Dorset, UK) medium supplemented with 10% foetal bovine serum (FBS: Life Technologies, Paisley, UK) and 2 mM L-glutamine (Life Technologies). Cells were grown in a humidified atmosphere with 5% CO2 in air at 37°C, and discarded every 6 weeks. For experiments with cannabinoids, the FBS was reduced to 5%. Authentication of the cell lines was performed by the service providers using the AmpFISTR Identifier Plus PCR amplification kit looking for the presence of <10 known loci for each cell line.
Cannabidiol (CBD) and Δ9-tetrahydrocannabinol (THC) were received from GW Pharmaceuticals Ltd. (Salisbury, UK) and were assessed in their purified (P) form, dissolved in ethanol. The final ethanol concentration in cell cultures were <0.1%. The cannabinoids used in these studies contained >96% of both CBD and THC respectively, however the compounds were prepared and used at the specified concentrations according to their total molecular weight rather than according to the specific weights of the active component within them.
RNA extraction and microarray analysis. Exponentially-growing cells were seeded into 6-well plates (BD Biosciences, Oxford, UK) at a concentration of 2×105 cells/well and were left to adhere overnight. Cells were then treated for 4 h with 10 μM of cannabinoid, either as CBD alone, THC alone or a combination of the two at a ratio of 1:1 (effectively 5 μM CBD and 5 μM THC). After this time, RNA was processed as described previously (13). Samples were processed for microarray analysis also according to methodologies detailed previously (13). Briefly, equal amounts of biotinylated cRNA was hybridised to the Illumina human HT12-v3 arrays for 18 h and subsequently processed according to the manufacturer's instructions before scanning on an Illumina BeadArray Reader. The image data were processed using default values in GenomeStudio v2009.1 with imputation of missing data, before loading onto GeneSpring v9.0 for data normalisation and filtering. A greater than 0.25-fold change was used as our cut-off magnitude for gene list compositions by using Excel software.
Immunoblot analysis. Cells were seeded into 6-well plates at a density of 2×105 cells/well and left to adhere overnight. The following day they were then treated with the specified compounds for 4 h, unless otherwise stated. Cells were then harvested by scraping into lysis buffer (New England Biolabs, Hitchin, UK), and standard western blot protocols were followed as described previously (14). Primary antibody probing was performed with anti-HSP40, HSP60, HSP70, HSP90, and cdk1 (all New England Biolabs). Anti-GAPDH (New England Biolabs) was used as a loading control. All antibodies were used at a dilution of 1:1000, followed by the appropriate HRP conjugated secondary antibodies (New England Biolabs) also at a dilution of 1:1,000. Bands were visualised by the SuperSignal chemiluminescent detection system (Thermo Scientific, Northumberland, UK). Densitometry of band intensity was determined using Image J software (NIH, Bethesda, MD, USA). Loading errors were controlled for using the band densities of respective loading controls and all samples were then expressed relative to this.
Assessment of intracellular reactive oxygen species. Exponentially-growing cells were seeded into 6-well plates at a density of 2×105 cells/well, left to adhere, and then incubated with 10 μM 2’,7’-Dichlorodihydrofluorescein diacetate (DCFH-DA: Sigma Aldrich) for 1 h. After this time, cells were washed twice with PBS and returned into fresh culture medium. Cells were then treated for 24 h with 10 μM CBD, 10 μM THC or 5 μM of CBD and THC concomitantly, both in the presence and absence of 10 mM N-acetyl cysteine (NAC: Sigma Aldrich). Cells were then harvested using trypsin, washed twice and re-suspended in PBS. DCFH-DA is a non-fluorescent reagent that is rapidly converted into the fluorescent product dichlorofluorescein by oxidation. We, therefore, used fluorescence as a marker of ROS presence, which was assessed flow cytometrically using a Becton Dickinson FACSCalibur (BD Biosciences). Data was gated on fluorescence width and area to remove doublet artefacts and to discriminate cells from debris. Mean fluorescence intensity for each sample was determined using the non-proprietary cell cycle analysis program WinMDI v2.9 (http://facs.scripps.edu/software.html).
Proteomic analysis of cellular supernatant. U87MG cells were seeded into 6-well plates in 1 ml of culture medium at a concentration of 2×105 cells/well and left to adhere overnight. Cells were then treated for 48 h with either 10 μM CBD, 10 μM THC or 10 μM CBD with 10 mM NAC. Supernatants were then collected and spun briefly at 1,000 rpm to remove large cellular debris, and the profile of 26 proteins associated with cellular stress was established by a proprietary assay kit according to instructions (Human Cell Stress Assay kit; R & D Systems, Abingdon, UK). The chemiluminescent signal was then visualised using the SuperSignal detection system, and capture spot density for each analyte expressed relative to control spots using the ImageJ software.
Potentiation of cytotoxicity assays. Exponentially-growing cells were added to 96-well plates at a density of 2,000 cells/well and left to adhere for 24 h. Dose response curves of CBD were generated both in the presence and absence of an inhibitor of HSP; either HSP90 using 17-allylamino-demethoxygeldamycin (17AAG) (Melford Biolaboratories Ltd, Ipswich, UK) or HSP70 with VER155008 (Insight Biotechnology, Wembley, UK). Cells were incubated with increasing concentrations of CBD (0.1-100 μM) and with fixed concentrations (1/2×IC50, 1×IC50, 2× IC50) of the HSP inhibitors, ensuring an equal total volume of 200 μl across the plate. Cell viability was assessed using a methylthiazoletetrazolium (MTT)-based assay according to methods previously described (15) using a GloMax Multi Plus microplate reader (absorbance: 560nm) (Promega, Southampton, UK). Data was analysed using GraphPad Prism curve fitting software (La Jolla, CA, USA).
Clonogenic survival assays. Exponentially growing cells were seeded into small flasks at a density of 2×105 cells/flask and left to adhere overnight. Cells were then exposed to CBD for 4 h, both alone and in the presence of the HSP inhibitors 17AAG and VER155008. After this time, cells were irradiated with 0-5 Gy using a 137Cs cell irradiator (IBL437C; CIS Bio International, France). Clonogenicity of the cells were then assessed as described previously, and the effect of the HSP inhibitors on the efficacy of CBD as a radio-sensitiser was calculated by determining the sensitiser enhancement ratios at 10% survival (SER10) (14).
Results
Target identification by surveying the transcriptome – HSPs. Standard microarray analyses of glioma cells treated with CBD-alone, THC-alone or a combination of the two were performed as a way of identifying novel targets for the cannabinoids. Comparing the number of genes that were altered following the different treatments showed that the types of genes or the genetic profile of the effects were cannabinoid-specific, and there was little overlap in the genes affected. Unsupervised assessments of the genes based upon rank fold-changes revealed a proportion of these genes were associated with HSPs (Table I). Specifically, examination of the top 25 genes up-regulated in T98G following treatment with CBD alone showed that 24% (6) genes were HSPs. We focussed on this by comparing the effects that these treatments had specifically on HSP transcripts, and showed that CBD tended to increase the expressions, whilst THC generally did not (Table II). For example, out of the 29 HSP-related genes called present in at least one treatment condition in T98G cells (Table II) 76% (22/29) were up-regulated following treatment with CBD compared to 28% (8/29) after THC. Full gene lists are available at ArrayExpress (www.ebi.ac.uk – accession number E-MTAB-3666).
CBD alters the expression of HSPs. To confirm the effects that CBD has on the expression of a number of HSP transcripts, western blots were performed using whole cellular lysates from cell lines cultured with CBD, THC or an equal combination of both. Results were similar in both T98G and U87MG cells so only blots from the latter are presented. There were duration and dose-dependent increases in HSP 40, 60, 70 and 90 (Figure 1a). In correspondence to the genomic data, THC had no remarkable effects on the HSPs studied, whilst the CBD/THC combination caused some increases in them at the longer durations (Figure 1b). The modifications to HSPs by CBD were not seen when other drugs were tested, and by and large, the other common chemotherapy agents that were used at equipotent concentrations (~IC50) did not increase the expression of the HSPs tested (Figure 1c). Additionally, the effects of CBD did not alter HSP expressions in another common cell line HCT116 (Figure 1d).
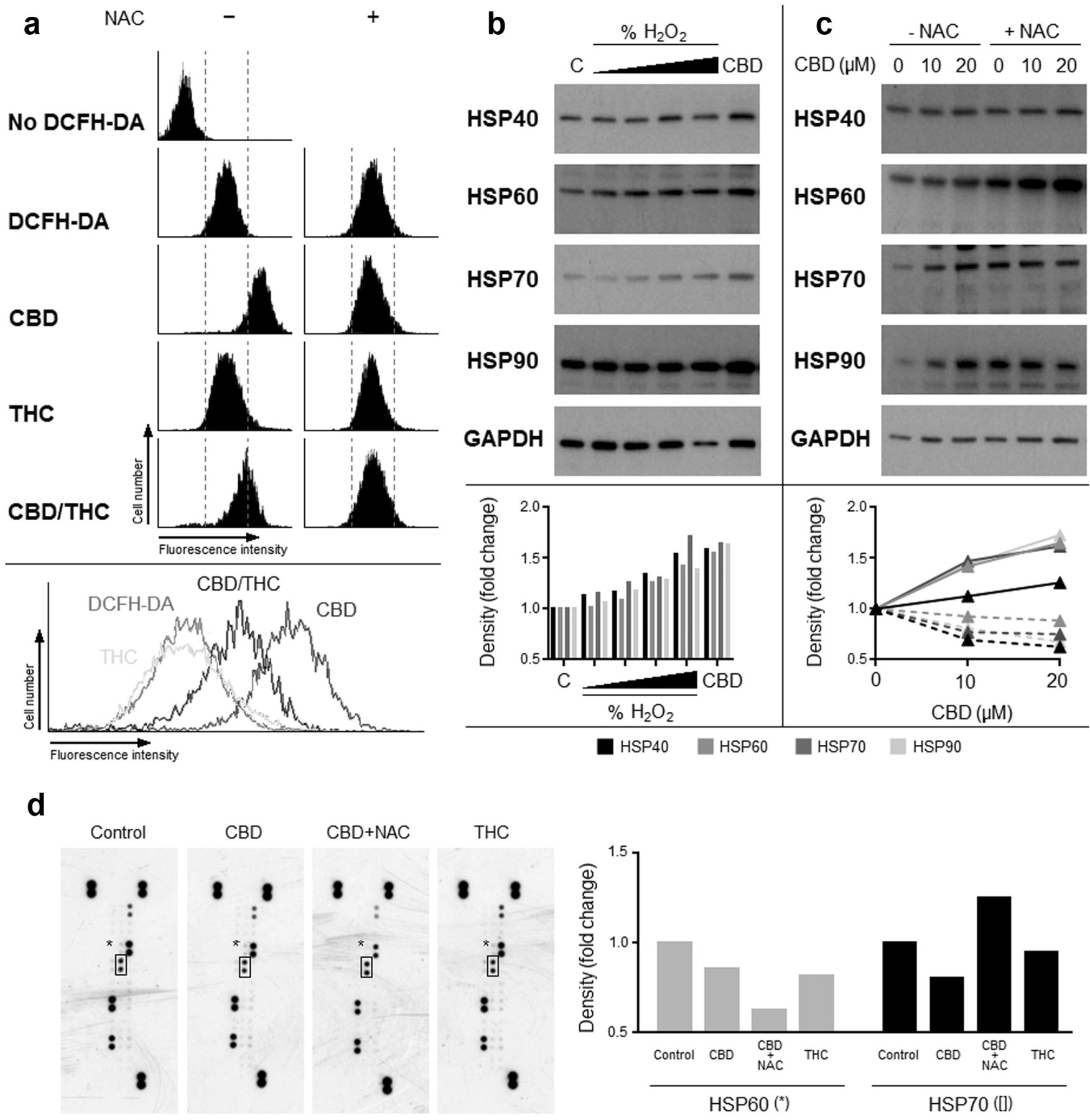
CBD generates oxidative stress that leads to an up-regulation of HSPs. In an attempt to understand the relationship between CBD and the up-regulation in HSPs, we next explored the effects that CBD may have on oxidative stress, as this has been shown to be a regulator of HSP expression. CBD metabolism in cells can stimulate the production of reactive oxygen species (ROS) that have a cytotoxic effect, and our results were in agreement with this and showed that CBD treatment resulted in a higher level of ROS production (Figure 2a). This was shown by assessing the intensity of a fluorescent product that is converted from the optically-inert substrate DCFH-DA by ROS. The ROS scavenger NAC reduced the fluorescence and suggested the de novo generation of ROS. THC did not appear to stimulate the production of ROS, whilst the mixture of CBD and THC produced an intermediate amount of ROS, which was negated by NAC (Figure 2a).
Top 25 genes induced by treatment with cannabinoids.
Having shown ROS production was increased following CBD, we next studied the effect that this may have on the expression of HSPs. The underlying rationale was the intrinsic compensatory response of the cell to increasing levels of HSPs as a means to counteracting ROS. Hydrogen peroxide was included in these studies as a positive control, and results showed that CBD significantly enhanced the expression of HSP 40, 60, 70 and 90 (Figure 2b). Furthermore, increases in HSPs were prevented by the addition of NAC, which supported the importance of ROS in this action (Figure 2c). As there was an apparent increase in HSPs, we next explored the possibility that they may be exuded by cells as primitive forms of intracellular communications (16), and therefore exploited as putative biomarkers of drug action. The results of the spotted array indicated no significant changes to HSP60 or HSP70 levels in tumour supernatants (Figure 2d).
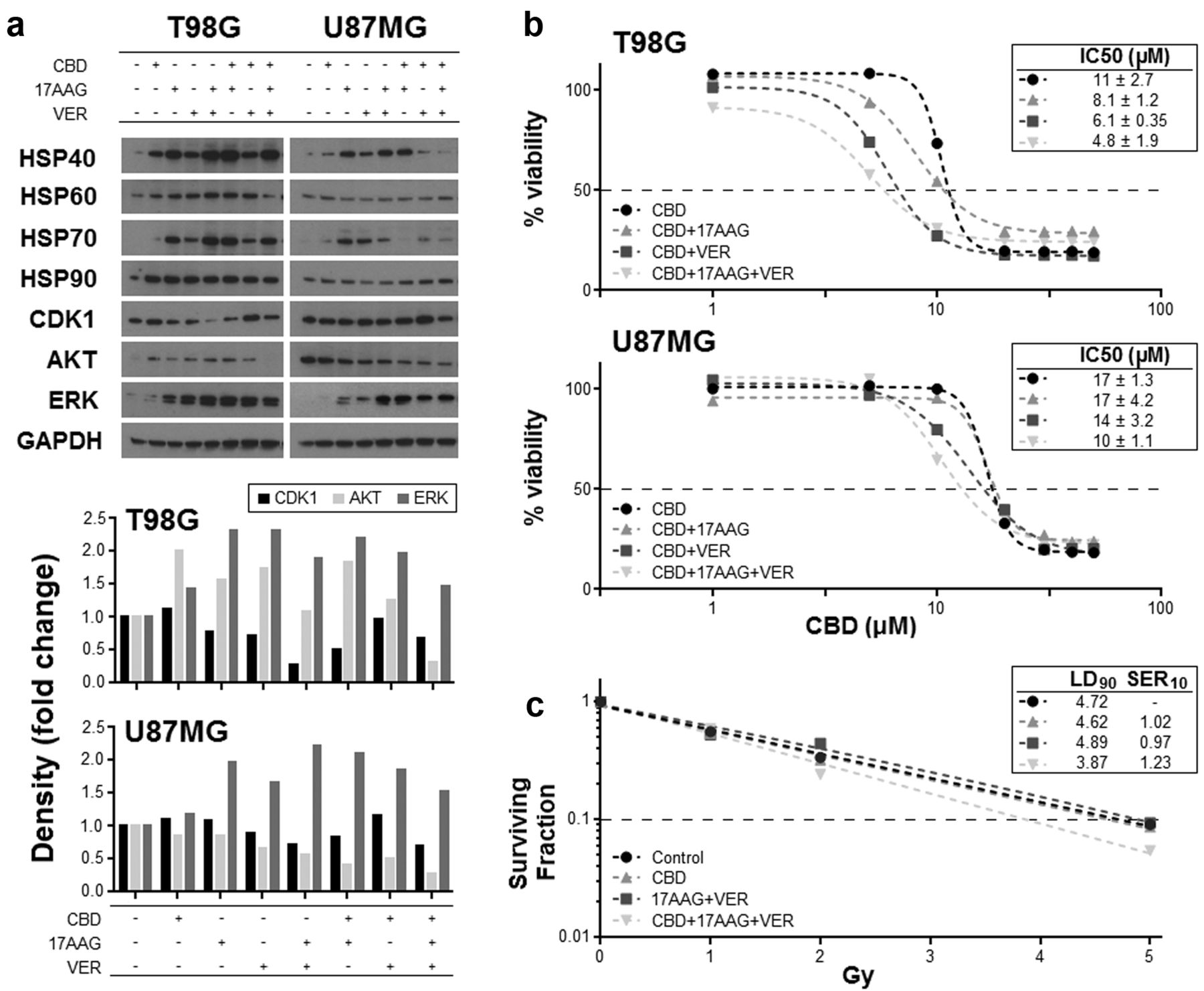
HSP inhibitors improve the efficacy of CBD. HSPs are principally chaperone proteins that shuttle client proteins to cellular systems that serve to amend crucial house-keeping proteins that are damaged by external stresses. These proteins vary depending upon the HSP in question, but crucially can include those that maintain the cancer phenotype. Similarly, HSPs can by dint of their sequestering ability, prevent pro-apoptotic proteins from performing their intended cytotoxic tasks. Consequently, inhibiting this function has been an aim of a number of experimental therapies. We then considered whether the increase in HSPs following CBD treatment in glioma cells could actually be preventing a better response from occurring. For this reason we co-cultured T98G and U87MG cells with CBD and an inhibitor of HSP70 (17AAG) or HSP90 (VER155008). These inhibitors were used at sub-cytotoxic concentrations, and their effects in modifying the IC50 of CBD were assessed by MTT assays.
Effect of cannabinoids on HSP genes in glioma cells.

Effect of cannabinoids on HSP protein levels. U87MG cells were cultured with CBD (0.5-20 μM) for up to 24 h. Cell lysates were prepared for determination of the effects of duration and concentration on HSP 40, 60, 70 and 90 levels by western blotting (a). The effect of treatments with THC alone or in combination with CBD and THC were also studied (b), as were the effects of common chemotherapy. Each one was used at equipotent concentrations (IC50) for 4 h (c). (C: control; E: ethanol control; GEM: gemcitabine; OXP; oxaliplatin; CPT: camptothecin; CPM: cyclophosphamide; LEN: lenalidomide; NTX: naltrexone). A second cell line (HCT116) was treated with CBD (0.5-20 μM) for up to 24 h before assessment of HSPs (d). Blots are representative of three separate experiments.
We first assessed the effects of 17AAG and VER155008 in glioma cells, and showed that they efficiently reduced the levels of the key client proteins AKT, ERK and CDK1 (Figure 3a). Treatment with CBD alone resulted in increases to these proteins, whilst the concomitant use of CBD with either one or both of the HSP inhibitors resulted in the reduction in the increased expression seen when CBD was used alone, suggesting the successful inhibition of the HSP (Figure 3a). Associated with these reductions in proteins were increases in the expression of HSP70 and HSP90, which has been reported previously as a compensatory response to the inhibition. Next, the effect of these treatments on cell number was assessed by MTT analysis. The increase in AKT and ERK as a result of increased HSP levels may be dampening cell death, and so the effect of negating these increases on the cytotoxic effect of CBD was evaluated by MTT analysis. Results showed that there was a reduction in the IC50 for CBD when HSP was inhibited simultaneously using chemical inhibitors (Figure 3b). Additionally, the co-treatment of cells with CBD and both HSP inhibitors increased radiosensitivity in U87MG cells as indicated by a greater fall in the surviving fraction of cells (Figure 3c).

Effect of cannabinoids on the levels of ROS and HSP in U87MG cells. Cells were pre-loaded with DCFH-DA for 1 h prior to exposure to cannabinoids individually or in combination. ROS presence was indicated by the fluorescent product dichlorofluorescein that is detectable by flow cytometry. NAC was employed as a ROS scavenger that negated oxidation of DCFH-DA (a). CBD was capable of increasing HSPs to a similar level seen with treatment with the higher concentrations of hydrogen peroxide (H202) (b), and the increases in HSPs were cancelled by NAC (c). Solid lines: -NAC; dashed lines: +NAC. Supernatants from U87MG cells treated with cannabinoids +/- NAC were also collected for stress-protein profiling by spotted array technology (d). Spots represent a panel of stress related proteins, including HSP60 (*) and HSP70 ([]).
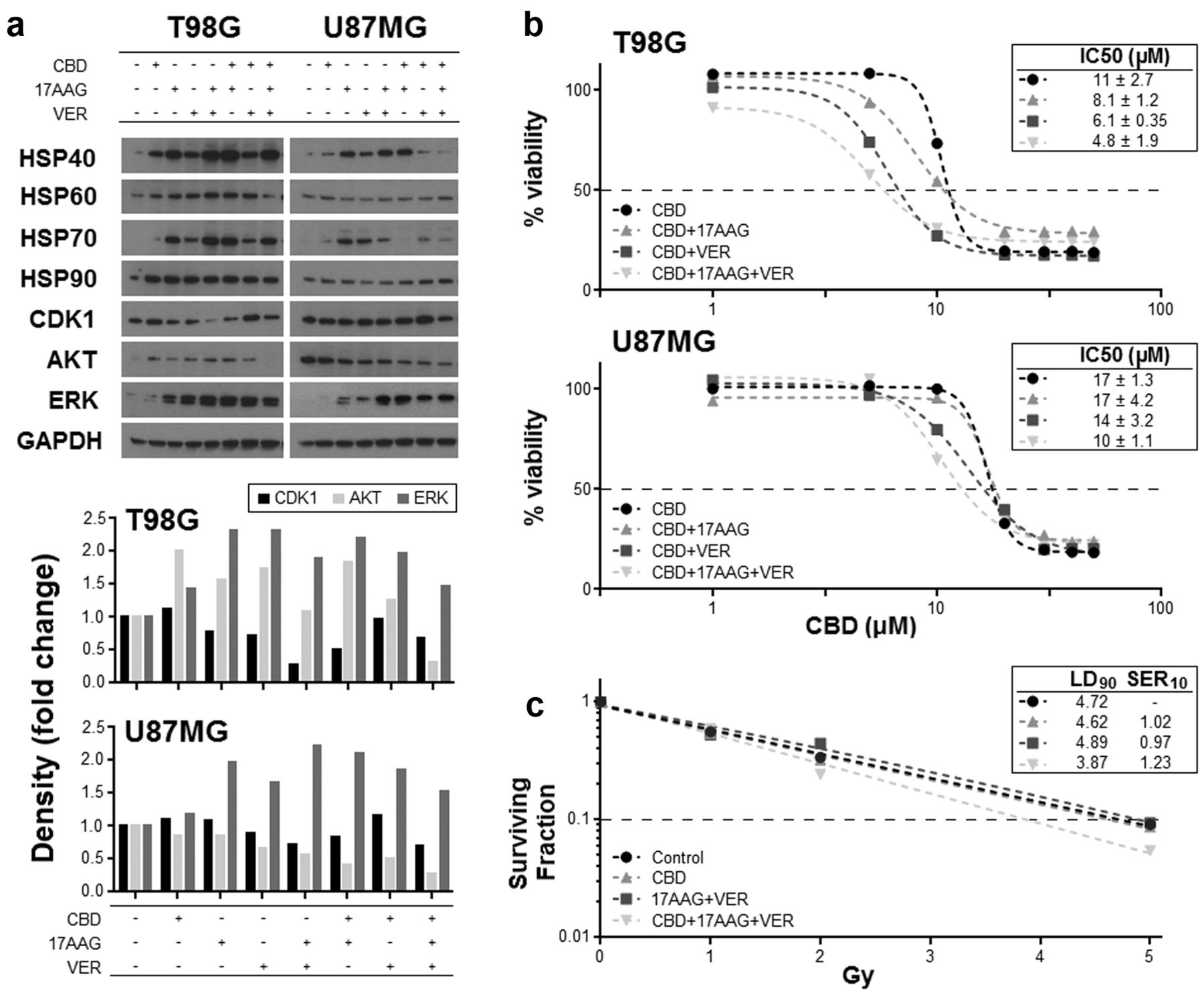
Effect of CBD and HSP inhibitors on HSPs and cell viability. U87MG and T98G cells were treated with 17AAG (1 μM) and/or VER155008 (VER: 20 μM) alone or with 10 μM CBD for 4 h. Lysates were collected and western blot techniques were employed to assess the levels of the HSPs and the client proteins CDK1, AKT and ERK (a). Densitometry was also performed and data shown represents the density of the treatment bands relative to their respective loading controls. Dose-response curves for CBD (48 h) were generated in U87MG and T98G cells concomitantly treated with sub-optimal concentrations of VER (20 μM) and 17AAG (1 μM) by using the MTT viability assay, and the effects of the HSP inhibitors on the IC50 values of CBD assessed (b). The effect that combining CBD with HSP inhibitors had on the sensitivity of U87MG cells to radiation was assessed by measuring the surviving fraction of cells following irradiation (c). Figures have been corrected for the minimal level of cell death caused by the HSP inhibitors when used alone.
Discussion
A number of active clinical trials in the UK and US testing the safety and/or action of cannabinoids are coming to fruition, and their outcomes are anticipated with interest. These trials were initiated following years of pre-clinical studies providing strong data/evidence of good biological activity, rendering the cannabinoids putative anticancer drugs. Studies have shown that these compounds can be successfully used both alone and in combination with other drugs, partly because cannabinoids have multiple modes of action. This increases the chance that they may interact favourably with other therapeutic modalities, and in accordance with this, we recently reported that the cannabinoids CBD and THC can be used together to successfully enhance the sensitivity of glioma cells to irradiation. Further understanding of the mechanisms of action of the cannabinoids, in particular the lead clinical candidates CBD and THC, is imperative as it will help guide new combinations and regimens, which would be useful clinically. Indeed, the ratio at which the cannabinoids are combined has been shown to be important to efficacy, as well as their schedule of administration and the introduction of a recovery phase in a pulse-like administration regimen (data not shown). Taken together, these factors highlight the intricate balance between the interactions of the cannabinoids and the importance of understanding their distinct mechanisms further. In an attempt to understand potential differences between the agents, the current study has focused on the impact that CBD has on heat shock protein biology. The significant finding of the study has been the way that pharmacologically inhibiting HSPs has enhanced the overall extent of cell death caused by CBD.
In the first part of the study we employed gene expression microarrays to identify transcripts involved in and/or associated with cannabinoid action. An unsupervised survey of the genes revealed that treatment with CBD, but not THC, was associated with an up-regulation of HSPs. The identification of HSPs as a ‘hit’ was further supported by the ensuing examination of all the HSP genes on the microarray chip, in samples that had been treated with CBD, which revealed the up-regulation of >70% of this cohort of genes in both cell lines. Western blotting analysis was used to confirm a general consensus between gene and protein expressions, which also demonstrated an increase in the HSP families following treatment with CBD. Conversely, these dramatic increases in gene and protein expression were not seen following treatment with THC or other common chemotherapy. This supports the possibility that the mechanisms of action for the cannabinoids are at some level distinct.
It remains unclear whether binding to the cannabinoid receptors, which are expressed on a range of tissue types, is a prerequisite for the activity of CBD or THC. Indeed, all cannabinoids have varying binding potencies for these receptors. Specifically, THC engages both the CB1 and CB2 cannabinoid receptors with high affinity (Ki values <100 nM), whilst CBD has virtually no affinity for either of them (Ki >2,000 nM) (17-19). This difference in affinity may explain the divergence in the action of the cannabinoids, especially in the context of anticancer action. Indeed, THC is thought to work via the CB1 receptor, causing the accumulation of members of the ceramide biosynthesis pathway, induction of cellular stress responses and initiation of autophagy and/or apoptosis (20-22). In contrast, CBD action is not dependent upon either of the cannabinoid receptors, or any other G-protein coupled receptors that have been conventionally linked to cannabinoid function, as well as not being associated with ceramide accumulation (23). Instead, the cytotoxic effects of CBD have been described to involve the production of ROS through direct interactions with mitochondria, and subsequent disruptions to intracellular signalling cascades, all resulting in the induction of apoptosis (24-26). Because CBD alters apoptotic mechanisms at the BCL-2/mitochondrial level (27), this may allow interactions/crosstalk between the pathways/mechanisms of apoptosis and autophagy to augment cell death (28, 29). It would be exciting to speculate that this is a putative hub of pathways through which CBD and THC work together to augment cell death.
Having identified HSPs in the arrays, we directed our attention more closely to the ability that CBD had in up-regulating ROS. ROS can affect HSP and are principally generated by mitochondrial respiration (30), but some intracellular organelles also display sufficient reducing power to generate them (31). This diverse group of molecules are generated by a variety of cells, especially those of the immune system, and can constitute part of the defence response to danger signals (32). In addition to this, other physiological roles have been ascribed to ROS, especially in the context of cancer (33). These include activation of signaling pathways (34, 35) and the direct damaging of DNA, which is in fact more persistent than standard irradiation (36, 37). By feeding into signaling pathways, ROS can influence the cellular systems that determine cell fate; indeed, the generation of ROS and their consequential effect on the release of apoptosis-initiating proteins is a way by which cell death can be brought about by these molecules (38). Our results confirmed the generation of ROS following culture with CBD, and likewise confirmed its absence after treatment with THC. We therefore directed our studies to examine further the relationship between ROS and HSPs, and the importance that these inter-related protagonists play in the cell killing actions of CBD in our cell types.
The JAK/STAT pathway is a signaling pathway that is activated by ROS, and whose members regulate a range of genes/proteins involved in cellular processes such as growth and death (35). One such group of proteins that are activated by the JAK/STAT pathway is the HSPs. Our studies showed that HSPs were increased as a consequence of ROS generation caused by exposure to CBD. In an attempt to deconvolute this relationship, we co-cultured cells with CBD and the ROS scavenger NAC in an attempt to neutralise the ROS. Results showed that NAC impeded the extent to which the HSPs involved with cytoplasmic and intracellular homeostasis, namely HSP70 and HSP90, were upregulated. Conversely however, the levels of the mitochondrial-specific HSP, HSP60, were increased in the presence of CBD and NAC. HSP60 is involved more with the chaperone of molecules involved with channelling proteins across the mitochondrial membranes, rather than with those involved with cell signalling (39). HSPs can be exuded by tumours and can thus act as putative adjuvants to toll-like receptors and in doing so stimulate immunity (40). This has led to investigations using HSPs as anticancer vaccines in patients with glioma (41). We therefore assessed the levels of HSPs exuded by the glioma cell lines following treatment with CBD, but showed them to be unremarkable.
Key client proteins of the HSPs are generally those involved in housekeeping, which include proteins that are necessary for conserving cell growth and survival such as CDK1, IKK and AKT (42). However, these proteins are also commonly dysregulated in cancer, and are the same proteins that can serve to maintain the cancer clone. One presumes that increasing their expression is undesirable, and in this regard, the upregulation of HSPs may be inadvertently damaging; in fact, HSPs are often over-expressed in cancer cells and have been modelled as a novel therapeutic target in cancer research (12). Our data showed that the CBD-mediated increase in HSPs resulted in higher levels of a number of client proteins, which was generally negated by the pharmacological inhibition of HSPs using 17AAG and/or VER155008. We next speculated that the increase in HSPs and their respective client proteins could unintentionally be blunting the anticancer effect of CBD due to their instinctive role in protection and survival. To explore this, we co-cultured glioma cells with a combination of CBD and the HSP inhibitors and then assessed cell viability. Results showed that reducing HSP upregulation brought about a greater degree of cytotoxicity. Specifically, the concentration of CBD required to reduce T98G cells by 50% (IC50) was 11±2.7 μM if used alone. However, concomitantly adding the HSP inhibitors at suboptimal doses (~IC20) into the cultures significantly reduced the IC50 of CBD to 4.8±1.9 μM. Similarly, we assessed the effect of the HSP inhibition on the lethality of irradiation, and showed cultures containing CBD and HSP inhibitors were superior in rendering cells sensitive to radiation.
In summary, these data describe a mechanism through which CBD can work to elicit an anticancer effect. Furthermore these data describe a method by which we can enhance the cytotoxic effects of CBD, by exploiting its known mechanisms of action and using new information to design successful combination strategies. Firstly we identified that HSP expression was altered following culture with CBD, and that this was an effect associated with the ability of CBD to increase the production of ROS by tumor cells. This is a mechanism of action that has been previously described by a number of groups; however, we have taken this further and described a reactive increase in HSPs which may inadvertently dampen the cytotoxic effects of CBD. We showed that including HSP inhibitors in cultures improved the ultimate cytotoxic nature of CBD, and that a chemotherapy and radiation treatment regimen comprising CBD with a pharmacological HSP inhibitor enhanced the impact of the irradiation modality. These studies thus provide us with a better understanding of how it is possible to enhance the use of this cannabinoid and improve its efficacy. This knowledge can be taken forward to the clinic where it can aid in the treatment of glioma.
Acknowledgements
The Authors wish to acknowledge the use of equipment located in the Medical Biomics Centre at St George's University of London. Full data sets for the gene data are available at ArrayExpress (www.ebi.ac.uk – accession number E-MTAB-3666).
Footnotes
Conflicts of Interest
This work was funded by a research grant from GW Pharmaceuticals Ltd., Salisbury, UK.
- Received July 7, 2015.
- Revision received August 10, 2015.
- Accepted August 13, 2015.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}