


Abstract
Metabolic reprogramming refers to the ability of cancer cells to alter their metabolism in order to support the increased energy request due to continuous growth, rapid proliferation, and other characteristics typical of neoplastic cells. This adaptation comprises of some important bioenergetic changes that, in contrary to conventional and outdated concepts, require an unchanged mitochondrial activity for their efficacy. This review highlights the characteristics of metabolic reprogramming, the role of mitochondrial activity in this particular setting, and the therapeutic possibility of targeting pathways of energy metabolism in cancer cells, with particular emphasis on metformin efficacy and its relationship with mitochondria metabolism.
Metabolic Reprogramming of Cancer Cells
Uncontrolled proliferation is one of the most relevant characteristics of cancer cells and not only does this main feature lead to the de-regulated control of cell proliferation but also corresponds to the adjustment of energy metabolism by ensuring the necessary energy is supplied as demanded in this particular condition (1). The most significant pathways involved in this adaptation seem to be aerobic glycolysis (2), glutaminolysis (3), and mitochondrial biogenesis and activities (4), all of which are increased in cancer.
In normal cells and under aerobic conditions, the metabolic activity primarily relies on the mitochondrial oxidative phosphorylation (OXPHOS) that generates energy through ATP (5). This is the most effective pathway under normoxic conditions, and is able to generate 36 molecules of ATP, since each glucose molecule is largely produced by mitochondrial respiration. In contrast, cancer cells become greedy for glucose because they preferentially use glycolysis for energy production, even under normoxic conditions (6). The activity of this pathway in oxygen abundance distinguishes it from normal anaerobic glycolysis, which is active in healthy cells under hypoxic conditions (7). Aerobic glycolysis produces only 2 molecules of ATP per glucose molecule and this limit can be exceeded by increasing the uptake of glucose through up-regulation of glucose transporters (8) (Figure 1). This characteristic is regularly exploited in positron-emission tomography imaging using a tracer to visualize tumors (9). Moreover, cancer cells increase the expression of several glycolytic proteins by regulating the balance between oncogenes and tumor-suppressor genes. Two oncogenes, namely MYC and hypoxia-inducible factor 1-alpha (HIF1A) are mainly involved (10). They are reported to be master inducers of cancer glycolysis, with a simultaneous lack of activity of p53, which ordinarily suppresses glucose uptake (10). The relationship between these factors increases the production of several key glycolytic enzymes, that strongly affect cancer-promoting glycolytic activity, as well as the general status of cancer metabolism (11). Glycolysis, in addition to energy, also provides cancer cells with biosynthesis precursors. Metabolites from this pathway are necessary for the synthesis of several macromolecules required for proliferation and other biosynthetic activities (12, 13).
Glucose is the principal energy source for cancer cells, although it is not the only one. Glutaminolysis (the process of glutamine catabolism) is a valid alternative pathway for mitochondrial energy production with ATP (14). Indeed, high glutamine consumption has frequently been observed in several cancers and glutamine metabolism supports both cancer cell viability and proliferation by providing energy and pools of Krebs cycle intermediates for biosynthesis of proteins, lipids, and nucleotides (8). Thus, similarly to glycolysis, glutaminolysis supports cancer cells by providing them with energy production and also with precursors necessary for biosynthesis (15). Moreover, glycolysis seems to be regulated by the activity of MYC and other oncogenes and tumor-suppressor genes (16).
It has long been believed that changes in cancer cell metabolism were independent of mitochondrial activity and that mitochondrial metabolism was inadequate to satisfy the energy demands typically present in rapidly proliferating cells. In addition, previous theories suggested that carcinogenesis was due to an impairment of mitochondrial activity (17). In particular, the transition of cellular metabolism to aerobic glycolysis, or the Warburg effect, suggested mitochondrial damage in cancer cells. Moreover, the mitochondrial production of reactive oxygen species (ROS), a recognized inducer of genomic instability, can lead to mitochondrial dysfunction as a metabolic hallmark of cancer cells. Conversely, according to recent investigations, mitochondria play a very important role in cancer development and progression, as demonstrated by the evidence that the function of mitochondria is intact in most cancer cells and that they generate the energy required by the tumor, just like in normal cells (18, 19). Recent studies provide substantial evidence that mitochondrial functionality and a healthy mitochondrial metabolism are essential for carcinogenesis. Mitochondria can support cancer cells through an indirect action, ROS-mediated action, or directly through mitochondrial biogenesis, which increases mitochondrial availability, since energy production also ensures the synthesis of many molecules indispensable for cellular biosynthesis, growth, and proliferation (20, 21). In particular, in hypoxia (where genetic repair processes are suppressed) there is a progressive increase in mitochondrial ROS production with a consequent increase in DNA mutations driving the malignant transformation process (22). With regard to direct mitochondrial action, cancer metabolic reprogramming has been previously explained by the metabolic shift from mitochondrial OXPHOS to aerobic glycolysis (the Warburg effect) (23, 24). This effect was originally explained as a result of a permanent impairment of mitochondrial activity (17). Recently, this view was challenged by investigations according to which the reduced efficiency of OXPHOS and altered functionality of mitochondrial activity are not common in cancer cells. Indeed, cancer cells frequently alter their mitochondrial function to switch from maximal energy production in order to meet the increasing demand typical of rapid cell proliferation (25). Moreover, c-MYC is also a strong promoter of mitochondrial synthesis and the possible lack of expression has a severely suppressive impact on many cancer metabolic pathways (26). Thus, mitochondrial biogenesis and activity are essential for the functionality of tumor cells, and impairment of this pathway could significantly suppress carcinogenesis and tumor growth, as well as explain the reason why mitochondria could be an excellent target for cancer therapy.
Molecules under investigation for their ability to inhibit mitochondrial activity and potential pathways.
Mitochondria and Cancer
Mitochondria are considered the power plant of the cell and their activity is crucial for energy production so that they maintain energy homeostasis in normal cells (27). As already mentioned, opposed to normal cells, required energy is produced by aerobic glycolysis in transformed cells and is also supported by mitochondrial activity in contrast with Warburg's opinion (28). Currently, it is substantially evident that cancer cells robustly engage in the two pathways, glycolysis and mitochondrial metabolism, in order to provide macromolecules with the energy that is essential for proliferation. However, the Warburg effect does not represent the fundamental difference between normal and cancer cells, rather it is the consequence of mitochondrial-dependent metabolic reprogramming in highly proliferating cells. Indeed, besides their role in energy production, mitochondria are also involved in apoptotic and autophagic cell death and have a close relationship with oncogenes, tumor-suppressor genes, and ROS production.
Mitochondria and apoptosis. Many studies have shown that mitochondrial activities play a pathogenic role in cancer development and progression, in particular as a consequence of fundamental regulation in cell death by apoptosis or necrosis. Mitochondria participate in apoptosis through different mechanisms, the most important of which seems to be the ability to release proteins, such as cytocrome c to promote caspase activation (29). Apoptosis, or programmed cell death, is a mechanism through which cells undergo suicide in order to control proliferation. It is an essential process required to maintain tissue homeostasis (30). Interestingly, stimuli triggering apoptosis converge on mitochondria that in normal cells release a series of apoptosis-inducing proteins (31). All perturbations of this pathway have an obvious role in the development of cancer and its progression, and the alteration of mitochondrial permeability or the inactivation of the pro-apoptotic members of the B-cell lymphoma gene-2 (BCL2) family are only some of the most striking examples (32). In cancer cells, the mechanism which disables apoptosis includes the contemporary loss of function of the pro-apoptosis tumor-suppressor gene p53 (33) and gain of function of the anti-apoptotic and oncogenic gene BCL2 (34). This occurs in several cancer types and is generally correlated with worse outcome (35). Different types of cellular stress can induce apoptosis through expression of glycolytic enzymes which are also highly expressed in cancer cells, in particular when mitochondrial membrane permeabilization (a part of the apoptosis pathway) does not work properly, thus suggesting a link between aerobic glycolysis and mitochondria-mediated apoptosis pathway (36).
Mitochondria and autophagy. Autophagy is a catabolic process that plays a pivotal role in cellular homeostasis (37). Indeed, it is able to degrade unnecessary or dysfunctional components deriving from cellular processes through the lysosomal pathway. As already mentioned, mitochondria generate decisive pathways regulating cell energy metabolism, as well as the balance between cellular life and death. Any alteration in mitochondrial homeostasis, including autophagy, is involved in the development of many diseases, including cancer. Mitophagy is a specialized form of autophagy in which mitochondria are specifically targeted for degradation (38). Interestingly, mitophagy seems to have a double role in carcinogenesis, probably depending on tumor type and stage. It seems to support survival and promote death in different cellular contexts. Generally, mitophagy aims to remove dysfunctional mitochondria to alleviate oxidative stress and prevent carcinogenesis. However, under adverse conditions, such as poor nutrient availability or hypoxia, it can protect cells from apoptosis and promote tumor cell survival (39). Mitophagy, therefore, seems to be a key quality-control factor in cancer cells and thanks to this feature, there might be a possibility of targeting this pathway for cancer intervention. Indeed, reduced mitochondrial metabolism deriving from inhibition of mitophagy may promote tumor cell death and its effect may be synergistic and further amplified by increased ROS. An alternative approach should combine inhibition of mitophagy with drugs able to induce mitochondrial stress signaling, such as metformin.
Oncogenes, tumor-suppressor genes and mitochondrial activity. Many oncogenes and tumor-suppressor genes are involved in metabolic reprogramming of cancer cells through a series of oncogenetic transformations (40). Although these transformations might be initially mitochondrial independent, recent evidence has shown that they are definitely involved in the regulation of mitochondrial metabolism and activity. Several oncogenes and tumor-suppressor genes also control cellular metabolism and contribute to the Warburg effect. MYC, phosphatidylinositol 3-kinase (PI3K) and HIFs have been implicated in enhanced glycolytic activity (41), while the tumor suppressor p53 has been shown to be critical for cell survival following glucose depletion (42). The activation of the PI3K/ serine/threonine kinase AKT (also known as protein kinase B or PKB) pathway, a feature commonly found in cancer cells, leads to enhanced glucose uptake and glycolysis and utilizes pathways relying upon functional mitochondrial metabolism (43). Hypoxia and oncogenes can alter tumor metabolism to support cell proliferation and carcinogenesis; HIF and, in particular, its subunit HIF1α are essential for the survival of transformed cells, playing a critical role in the regulation of glycolysis under hypoxic conditions (44). c-MYC was found to be amplified and constitutively active in several types of tissue and cancer. It is a classical transcription factor and it regulates cell proliferation and differentiation (45). The activation of this gene increases the level of glucose transporters as well as glycolytic enzymes, and in this field, c-MYC-induced metabolic changes mimic hypoxic effects under normal oxygen conditions. This demonstrates that it can facilitate aerobic glycolysis (46). Interestingly, c-MYC is able to induce the promotion of HIF expression with simultaneous inhibition of the degradation of the HIF1α subunit (47). c-MYC was recently shown to directly regulate mitochondrial DNA replication and consequently stimulate abnormal mitochondria in transformed cells (48). c-MYC also enhances the transcription of glutaminase-1 and up-regulates glutamase production and glutaminolysis, a valid alternative pathway for mitochondrial energy production with ATP (49).
p53 is one of the most important tumor-suppressor proteins and plays a fundamental role in several cellular functions, including the promotion of apoptosis, regulation of the cell cycle, senescence, and DNA repair, each of which works to prevent cancer development and progression (50). Obviously, every mutation or depletion of this protein is associated with the loss of its ability to hinder malignant disease (51). Recently, increasing evidence has shown that p53 plays an important role in the regulation of both glycolysis and OXPHOS (52). This protein generally promotes OXPHOS and dampens glycolysis, while alteration of p53, or its suppression, leads to a disrupted balance, with oncogenic transformation as a consequence. In spite of the fact that the genetic basis of the predominant glycolytic activity observed in cancer cells is likely to be the result of complex cooperation of several pathways, p53 was recently demonstrated to be able to regulate mitochondrial respiration with secondary changes in the glycolysis pathway (53). Finally, p53 was shown to repress the transcription of glucose transporters to inhibit glucose uptake and overall ROS production (54).

Metabolic activities in cancer (Warburg effect) and normal cells. OXPHOS, Oxidative phosphorylation; FADH2, flavina adenina dinucleotide; NADH, nicotinammide adenina dinucleotide; ATP, adenosina trifosfato; GLUT, glucose transporter Proteins.
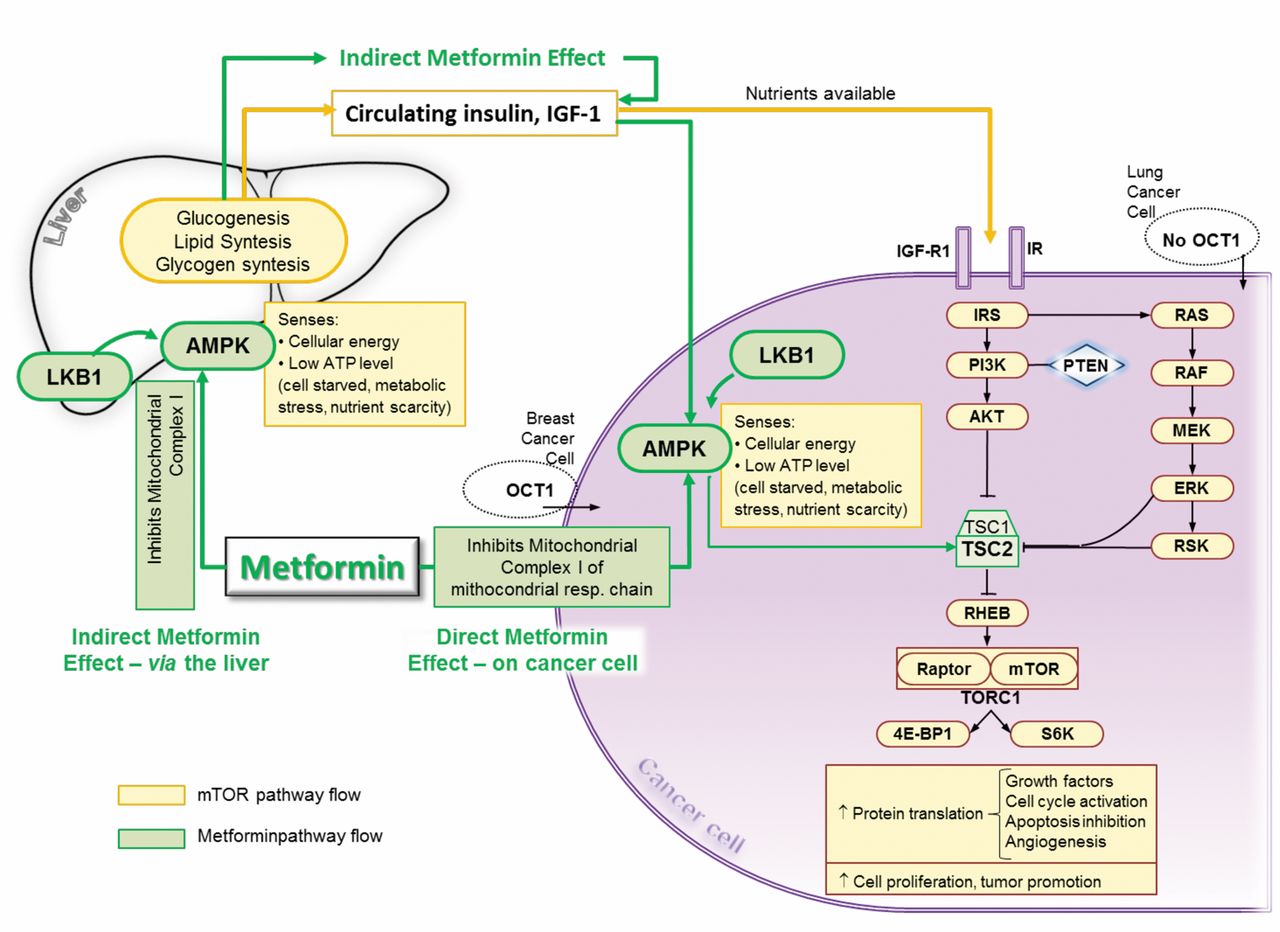
Mechanisms of action of metformin. AMPK, 5′ adenosine monophosphate-activated protein kinase; LKB1, liver kinase B1; ATP, adenosina trifosfato; mTOR, mammalian target of rapamycin; IGF-1, Insulin-like growth factor -1; IGF-R1, Insulin-like growth factor receptor 1; IR, insulin receptor; OCT1, organic cation transporter-1; PI3K, phosphatidylinositol 3-kinase); AKT, serine/threonine kinase; TSC1, tuberous sclerosis complex tumor suppressor gene 1; TSC2, TSC2, tuberous sclerosis complex tumor suppressor gene 2; PTEN, phosphatase and tensin homolog; RHEB, Ras homolog enriched in brain; TORC1, Transcriptional coactivator for CREB1; 4E-BP1, 4E binding protein 1; S6K, ribosomal protein S6K, S6 kinase; RAS, RAS gene super-family; RAF proto-oncogene serine/threonine-protein kinase; ERK, extracellular-signal-regulated kinases; Rsk, ribosomal s6 kinase.
Mitochondria and ROS. ROS includes a series of oxygen-derived molecules, historically considered responsible for several damaging effects (55). Oxidative stress is assumed to contribute to cancer and, in particular, to tumor development and growth, mainly through its ability to damage proteins, lipids and DNA and eventually produce genomic instability (56).
The major source of intracellular ROS is oxidative metabolism, mainly generated by the mitochondrial respiratory chain (57). These mitochondria-generated free radicals have important implications not only in inflammatory processes and a variety of other diseases, but also in cancer development, where they can cause initiation of mutations of mitochondrial DNA, promoting malignant transformation as well as metastatic behavior (58). Generally, under physiological conditions, mitochondria trigger redox signaling in the cell by releasing ROS. However, under pathophysiological conditions, they can also contribute to cancer promotion and amplify cancer cell phenotypes. These effects are obtained through their ability to regulate different signaling pathways. For example, mitochondrial ROS seem to affect the PI3K pathway (59), well-known for increasing proliferation, growth, and cellular survival. These effects are amplified by the presence of ROS (60). Mitochondrial ROS are also able to stabilize HIF, one of the most important mechanisms through which tumor cells adapt to the diminished oxygen conditions in the microenvironment (hypoxia) typical of cancer, and to promote survival and angiogenesis, favoring cellular metabolic reprogramming to increase glycolysis (61). ROS signaling promotes cancer proliferation and the ability to disrupt mitochondria-to-cell redox communication could be a promising target for therapy. There exists substantial evidence supporting the concept that reprogramming of cellular metabolism is a crucial aspect of cell transformation, resulting both from the involvement of several cellular pathways and from mutations in oncogenes and tumor-suppressor genes. This phenomenon is highly dependent on mitochondrial metabolism and activity.
Targeting Mitochondria: Mitochondrial Inhibitors
Mitochondrial metabolism has since long been considered irrelevant to the high proliferative rate typically exhibited by cancer cells; mitochondrial dysfunction was viewed as a metabolic hallmark of cancer cells. Contrary to this, mitochondrial metabolism was recently accepted as being an essential part of carcinogenesis and consequently an interesting target for cancer therapy. (19, 21). In spite of this, significant challenges still remain with regard to translating pre-clinical active drugs targeting mitochondrial metabolism. For example, a better understand over the biology of mitochondrial metabolism is mandatory, together with its relationship with carcinogenesis. This may lead to the development of active and safe therapies combining mitochondrial metabolic inhibitors and conventional or new anticancer agents.
We know that mitochondria can use different biochemical processes in order to produce ATP. In particular, the electron transport chain in the mitochondrial respiratory pathway is essential for OXPHOS in causing generation of ROS during oxygen contact. Thus, the balance between redox and generation of cellular ATP is essential for cellular proliferation and health. For these reasons, targeting the activities of bioenergetic mitochondrial activities using appropriate drugs is a promising therapeutic strategy to target cancer cells; although any drug targeting mitochondrial activity should be previously tested for its toxicity towards healthy cells, this approach remains highly possible and intriguing. There are several molecules under investigation regarding their ability to inhibit mitochondrial activity. Table I summarizes the drugs known to be active in mitochondrial functionality and the potentially involved pathway.
Some of the most promising molecules that seem to be effective on mitochondrial bioenergetics activities and considered as mitochondrial poisons are mainly the following: resveratrol, a polyphenolic compound, is able to inhibit mitochondrial ATP synthase and lead to 5′ adenosine monophosphate-activated protein kinase (AMPK) activation, also contributing to an insulin-sensitizing effect (62). Berberine, another herbal insulin sensitizer, participates in mitochondrial function suppression through AMPK activation, with consequent glucose and fatty acid oxidation (63). Epigallocatechin gallate stimulates mitochondrial biogenesis and promotes OXPHOS through a cAMP/protein kinase A (PKA) - and sirtuin-dependent mechanism (64). Its antitumor activity is achieved by the increase of apoptosis through mitochondrial damage and mitochondrial membrane depolarization (65). Moreover, curcumin, a phenolic compound, exhibits bifunctional antioxidant properties related to its capability to react directly with ROS and recently, it has been postulated that this compound has activity against mitochondrial dysfunction (66).
The above fact indicate that several approaches are under investigation to obtain selective destruction of cancer cells based on their mitochondrial activity. The most promising of these approaches involves the bioenergetic role of mitochondria and drugs potentially effective on this pathway.
Metformin and Mitochondria
Approximately 10 years ago, a pivotal article reported a relationship between the use of metformin, a drug for diabetes, and reduced cancer incidence. Thanks to such data, an important cancer research program was carried out which included almost 200 observational, preclinical and clinical trials (67, 68). The whole set of trials confirmed the ability of this treatment to reduce cancer incidence and progression, both in diabetic and non-diabetic patients.
Although the anticancer action of metformin is not yet well established, recent data show this agent to be a key regulator of cellular metabolism and its action can be divided into two separate pathways (Figure 2): a) an indirect effect including an action on metabolism and, in particular, on glucose metabolism, such as reduced concentration of glucose and insulin in the blood and therefore of their mitogenic activity; and b) a direct effect on tumor cells through organic cation transporters (OCTs), able to reduce activity of the mitochondrial respiration chain and ATP production, which, in turn, activates AMPK, regulating energy homeostasis (69).
Inside the cancer cell, metformin primarily targets mitochondria. Its effects on cellular mitochondrial metabolism are not thoroughly understood. However, this drug seems to work through the inhibition of mitochondrial complex I in the electron transport chain, suggesting its direct action on mitochondria (70). Metformin can inhibit mitochondrial ATP production, inducing AMPK activation and consequently reducing mammalian target of rapamycin (mTOR) activity, necessary for cell proliferation. Reduced ATP production can cause cellular death, particularly in the case of lower glycolytic energy production, which is a direct consequence of limited glucose availability. Cells treated with metformin display reduced glucose metabolism and, in general, an overall decrease in mitochondrial respiration (71).
Recent data suggest individual variations with regard to the efficacy of metformin and, in particular, in metformin metabolism and its interaction with cellular targets (72-74). The level of metformin within cells is determined by the relationship between uptake and expulsion mechanisms. The uptake mechanism is directly dependent on the mitochondrial membrane potential, although it requires OCTs that are expressed on the surface of cells of several tumor types, and which are likely to uptake and expel a wide range of proteins (75). However, not all tumor cells express OCTs. Therefore, metformin may not accumulate and inhibit mitochondrial complex I in all types of cancer. This partially explains the why metformin is not effective against all tumor types. In addition, recent data seem to show that the variability of metformin efficacy might also be determined by genetic polymorphisms related to OCTs (76). The results of these trials have shown that metformin reversibly inhibits mitochondrial complex I within cancer cells and reduces tumorigenesis and that it would be most effective under low glucose and oxygen conditions. A combination therapy with drugs limiting glucose uptake and glycolysis (PI3K inhibitors), or with agents able to drive cancer cells to low glucose and oxygen levels (anti-angiogenic inhibitors) should probably be more effective than metformin alone. There is therefore a need to design specific clinical trials on the use of metformin as an anticancer agent aimed at a selected population determined on the basis of the expression levels of OCTs and mitochondrial genes.
Conclusion
Identifying molecular mechanisms involved in carcinogenesis provides strong rationales for developing strategies for cancer prevention and treatment. Metabolic reprogramming is now considered a promising target in this setting and in particular, mitochondrial activity is being considered as a target for cancer therapy. Recent evidence suggests that several mitochondrial inhibitors and particularly metformin, a widely used anti-diabetic drug, should be promising anticancer agents able to target mitochondrial energy production directly, limiting respiration through the inhibition of mitochondrial complex I. The efficacy of this approach should be considered ideal for reversing drug resistance in oncological patients for whom conventional therapies failed, using combination treatment of metformin with conventional other anticancer agents. Finally, this approach can lead the way to unlocking new anticancer therapies. Many questions remain open with regard to the translational pathway. It is mandatory to understand more on mitochondrial metabolism in carcinogenesis and it is also fundamental to establish the toxicity of metformin treatment in normal cells and in non-diabetics or in individuals with a healthy metabolism.
- Received July 8, 2015.
- Revision received September 1, 2015.
- Accepted September 10, 2015.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Why the tumor cell metabolism is not that abnormal
- Relationship Between Metabolic Disorders and Breast Cancer Incidence and Outcomes. Is There a Preventive and Therapeutic Role for Berberine?
- Suppression of Kaposi's Sarcoma-Associated Herpesvirus Infection and Replication by 5'-AMP-Activated Protein Kinase