


Abstract
Background: Long noncoding RNA ANRIL (antisense non-coding RNA in the INK4 locus) represses p15 and p16, which induce cell-cycle arrest at G1 phase, leading to enhanced cell proliferation of normal fibroblasts. Herein we report that ANRIL is also involved in the regulation of cancer-cell proliferation. Materials and Methods: HeLa and H1299 cells were transfected with ANRIL siRNAs. At 72 h post-transfection, cells were subjected to quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and cell-cycle analysis. Results: qRT-PCR showed that ANRIL is highly expressed in these cancer cells compared to normal fibroblasts. Depletion of ANRIL increased p15 expression, with no impact on p16 or ARF (alternative reading frame) expression, and caused cell-cycle arrest at the G2/M phase, leading to inhibition of proliferation of H1299 and HeLa cells. Conclusion: ANRIL positively regulates the proliferation of cancer cells, such as H1299 and HeLa cells, via regulating p15 and other genes related to G2/M phase control.
Antisense non-coding RNA in the INK4 (inhibitors of cyclin dependent kinase 4) locus (ANRIL) is a long noncoding RNA (lncRNA) located in the human chromosome 9p21 region. The INK4 locus encodes three tumor-suppressor genes: CDK (cyclin dependent kinase) inhibitors (CKIs) p15 and p16, and ARF (alternative reading frame), which is a positive regulator of p53. This region is frequently mutated or its expression silenced in human cancer (1-4). CKIs bind to and inhibit the activity of specific cyclin–CDK complexes, preventing G1 to S transition of the cell-cycle (5, 6). Among these CKIs, p15 and p16 are induced by a variety of oncogenes, such as activating RAS (rat sarcoma) mutants, causing stable cell-cycle arrest through inhibiting cyclin D-dependent CDK4/6 activity (7-9). ARF is also induced by various oncogenes, such as c-MYC (cellular myelocytomatosis oncogene), and antagonizes the activity of MDM2 (transformed mouse 3T3 cell double minute 2) ubiquitin ligase, thereby stabilizing p53 tumour suppressor protein and causing cell-cycle arrest (10-13). The activation of the INK4 locus is therefore important to protect cells from hyper-proliferative stimulation induced by oncogenic insults.
The ANRIL promoter is located between p15 and ARF, and ANRIL is transcribed in the antisense direction with these genes (14). Yap et al. (15) and our group (16) reported that ANRIL is involved in the transcriptional repression of the INK4 locus (15, 16). Inhibition of ANRIL in human normal fibroblasts increases the expression of p15 and p16, leading to a decrease in cell proliferation. Yap et al. showed that ANRIL associates with CBX7 (chromobox 7), a component of polycomb repression complex (PRC)-1 (15). We also showed that ANRIL associates with SUZ12 (suppressor of zeste 12 homolog), a component of PRC-2 (16). Inhibition of ANRIL disrupts the binding of PRC-1 and -2 on the INK4 locus, indicating that ANRIL recruits PRC-1 and -2 on the INK4 locus, leading to the repression of p15 and p16 transcription (15, 16). ANRIL is also involved in the occupancy of PRC-1 and -2 on other genes located on different chromosomes, indicating that it functions on different chromosomes to regulate target genes in trans (17). The trans-regulation by ANRIL is dependent on its Alu motif (17). Several studies showed that depletion or overexpression of ANRIL causes changes in the expression levels of many genes involved in cell proliferation, cell adhesion, gene expression, and apoptosis, suggesting that ANRIL is involved in multiple cellular functions (17-19). In the present study, we investigated the role of ANRIL in the proliferation of cancer cells, namely human non-small cell lung cancer H1299 cells and HeLa cervical cancer cells.
Materials and Methods
Cell culture. All cell lines used in this study were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA) containing 10% foetal bovine serum (GIBCO, Grand Island, NY, USA) and incubated at 37°C in an atmosphere containing 5% CO2.
RNA interference. siRNA oligonucleotides against ANRIL (SIGMA-ALDRICH, Tokyo, Japan) were transfected into HeLa and H1299 cells using Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer's instructions. HeLa and H1299 cells were incubated 72 h after transfection before analyses. The nucleotide sequence of ANRIL siRNA was 5’-GGUCAUCUCAUUGCUCUAU-3’ with 3’ dTdT overlaps.
Reverse transcription-polymerase chain reaction (RT-PCR) and quantitative RT-PCR (qRT-PCR). Total RNA was isolated by the RNeasy Plus kit (Qiagen, Tokyo, Japan) according to the manufacturer's instructions. The isolated total RNA was reversed transcribed into cDNA using SuperScript Reverse Transcriptase II (Invitrogen). For qualitative PCR, the produced cDNA was amplified by the specific primer sets: ANRIL, 5’-TGCTCTATCCGCC AATCAGG-3’ and 5’-GGGCCTCAGTGGCACATACC-3’; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5’-GCAAATTCCATGGCACCGT-3’ and 5’-TCGCCCCACTTGATT TTGG-3’; ribosomal protein L32 (RPL32), 5’-GGCGGAAACCCAGAGGCATTGA-3’ and 5’-CCTGGCATTGGGGTTGGTGACTCT-3’. For qRT-PCR, the produced cDNA was added to SYBR Green PCR master mix (Qiagen) and amplify by the specific primer sets: p16, 5’-CGGTCGGAGGCCGATCCAG-3’ and 5’-GCGCCGTGGAGCAGCAGCAGCT-3’; p15, 5’-AAGCTGAGCCCAGGTCTCCTA-3’ and 5’-CCACCGTTGGCCGTAAACT-3’; ARF, 5’-CCCTCGTGCTGATGCTACTG-3’ and 5’-ACCTGGTCTTCTAGGAAGCGG-3’; GAPDH, 5’-GCAAATTCCATGGCAC CGT-3’ and 5’-TCGCCCCACTTGATTTTGG-3’. Assays were performed in triplicate on a Mx3000P Real-Time Q-PCR System (Agilent Technologies, Santa Clara, CA, USA).
Cell-cycle analysis. HeLa cells were fixed overnight in 70% ethanol at −20°C. The DNA of fixed cells was stained by Muse™ Cell Cycle Kit (Merck Millipore, Darmstadt, Germany) according to the manufacturer's instructions. Cells were analyzed by Muse™ Cell Analyzer and analysis software (Merck Millipore).
Results
We first examined the expression of ANRIL in various human cell lines. RT-PCR assay showed that ANRIL was highly expressed in several cancer cell types, including ABC-1, H1299 (human non-small cell lung cancer), HeLa (cervical cancer) and Saos-2 cells (osteosarcoma) compared with WI38 and TIG-3 cells (normal diploid foetal lung fibroblasts) (Figure 1).
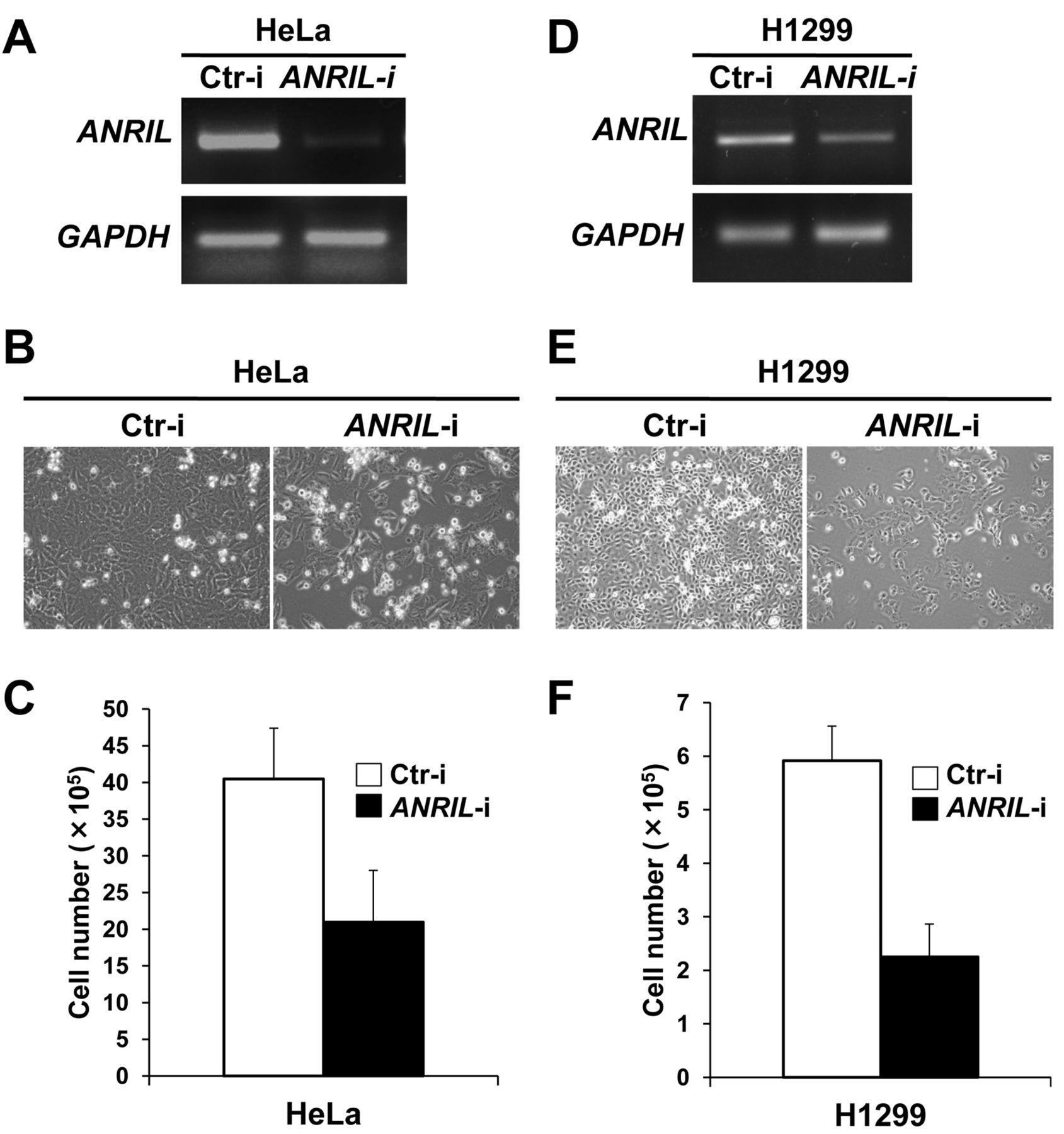
We previously reported that ANRIL positively regulates the proliferation of human normal diploid foetal lung fibroblasts (16). Therefore, we next determined whether ANRIL is also involved in the regulation of cancer-cell proliferation. We knocked-down ANRIL using specific siRNA oligonucleotides in cancer cells with high expression of ANRIL, namely HeLa and H1299 cells. RT-PCR assay confirmed that these siRNAs reduced ANRIL expression to nearly undetectable levels in HeLa cells (Figure 2A). The depletion of ANRIL resulted in repression of HeLa cell proliferation (Figure 2B and C). In H1299 cells with knocked-down ANRIL, we also observed a substantial decrease of H1299 cell proliferation (Figure 2D-F). These results suggest that ANRIL positively regulates the proliferation of HeLa and H1299 cells.

Expression levels of ANRIL (antisense non-coding RNA in the INK4 locus) in various human cells. The levels of ANRIL expression were determined by RT-PCR. RPL32 (ribosomal protein L32) was used as an internal control.
Yap et al. (15) and our group (16) previously reported that ANRIL is involved in the repression of p15 and p16 transcription. We, therefore, examined the effect of ANRIL silencing on the expression of genes of the INK4 locus including p15, p16, and ARF in HeLa and H1299 cells. qRT-PCR showed that silencing ANRIL in HeLa cells resulted in an about two-fold increase in p15 mRNA, with no changes in p16 or ARF mRNA levels (Figure 3A). Silencing ANRIL in H1299 cells also resulted in a more than fourfold increase in p15 mRNA, but no changes were observed for ARF mRNA (p16 was not detected) (Figure 3B). These results suggest that ANRIL is involved in p15 repression in cancer cells, such as HeLa and H1299 cells.
Given that silencing ANRIL increases the mRNA level of p15, which causes G1 phase arrest, we next examined the effect of silencing ANRIL on the cell cycle using Muse™Cell Analyzer. Contrary to our expectation, silencing of ANRIL in HeLa cells induced a pronounced G2/M phase accumulation compared with control cells (Figure 4A and B), suggesting that ANRIL regulates the G2/M phase of the cell cycle in HeLa cells.
Discussion
Recent studies revealed that several lncRNAs have pivotal roles in critical cellular processes, including proliferation, differentiation, apoptosis, and senescence (20-23). ANRIL is involved in the regulation of cellular senescence of normal human diploid foetal lung fibroblasts through the regulation of p15 and p16 transcription (15, 16). In this study, we showed that ANRIL is also involved in the regulation of proliferation of cancer cells, such as the human H1299 non-small cell lung cancer cells and HeLa cervical cancer cells. Silencing ANRIL increased p15 expression, but not p16 and ARF, suggesting that ANRIL represses mainly p15 transcription in these cancer cells. p15 inhibits the activity of cyclin D–CDK4/6, causing cell-cycle arrest at the G1 phase. Interestingly, silencing ANRIL caused cell-cycle arrest at G2/M phase in these cancer cells, not G1 phase. Recent reports demonstrated that ANRIL recruits PRC-1 and -2 to target genes located on different chromosomes to regulate gene expression (17). In support of this notion, silencing ANRIL impacts the expression of a large number of genes (18, 19). Thus, ANRIL may be involved in the regulation of genes that control G2/M phase in a trans-acting manner.

Silencing of ANRIL (antisense non-coding RNA in the INK4 locus) represses the proliferation of HeLa and H1299 cells. A: HeLa cells were transfected with control (Ctr-i) or ANRIL siRNA oligonucleotides. At 72 h after transfection, cells were harvested and subjected to RT-PCR (reverse transcription-polymerase chain reaction) to determine the level of ANRIL. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as an internal control. B: HeLa cells transfected with control or ANRIL siRNA oligonucleotides were observed by phase-contrast microscopy at 72 h after transfection. C: After initial seeding of 5×105 cells, HeLa cells were incubated overnight, and then transfected with control or ANRIL siRNA oligonucleotides. At 72 h after transfection, viable HeLa cells were counted by Trypan Blue staining. D: H1299 cells were transfected with control (Ctr) or ANRIL siRNA oligonucleotides. At 72 h after transfection, cells were harvested and subjected to RT-PCR as in (A). E: H1299 cells transfected with control or ANRIL siRNA oligonucleotides were observed as in (B). F: After initial seeding of 1×105 cells, H1299 cells were incubated overnight, and then transfected with control or ANRIL siRNA oligonucleotides. Viable H1299 cells were counted as in (C).

Silencing of ANRIL (antisense non-coding RNA in the INK4 locus) increases p15 mRNA expression. A: HeLa cells were transfected with control (Ctr-i) or ANRIL siRNA oligonucleotides. At 72 h after transfection, cells were harvested. The effects of ANRIL silencing on the expression of p15, p16 and ARF (alternative reading frame) were determined by qRT-PCR. The results are expressed relative to the corresponding values for control cells. The mean values and standard deviations were calculated from the data of three independent experiments. B: The effects of ANRIL silencing on the expression of p15 and ARF in H1299 cells were determined by qRT-PCR as in (A).

Silencing of ANRIL (antisense non-coding RNA in the INK4 locus) causes cell-cycle arrest at the G2/M phase. A: HeLa cells transfected with control (Ctr-i) or ANRIL siRNA oligonucleotides were analyzed by Muse™ Cell Analyzer at 72 h after transfection. B: The percentage of total cells present in the G0/G1, S and G2/M phases of the cell cycle are shown.
We also showed that ANRIL is highly expressed in different types of human cancer cell, such as non-small cell lung cancer (ABC-1 and H1299), cervical cancer (HeLa) and osteosarcoma (Saos-2). High levels of ANRIL have been previously observed in human cancer, including prostate (16) and gastric (24). The molecular mechanism underlying aberrant expression of ANRIL in cancer remains to be elucidated. It was reported that transcription factor E2F1 induced by ATM (ataxia telangiectasia mutated) binds to the ANRIL promoter and activates its transcription in response to DNA damage (18, 25). Increased expression of E2F1 has been observed in many types of human cancer (26), suggesting that deregulation of E2F1 might lead to aberrant expression of ANRIL in cancer.
p15 and p16 are activated by oncogenes, including an oncogenic form of small GTPase RAS (called oncogenic RAS), causing stable cell-cycle arrest in order to protect cells from hyper-proliferation (7-9). Recently, we reported that oncogenic RAS reduces the expression of ANRIL (15, 27). The decrease of ANRIL by oncogenic RAS might be required for p15- and p16-dependent cell-cycle arrest. We, therefore, postulate that aberrant expression of ANRIL might disrupt protection by p15 and p16, leading to oncogenic transformation.
Acknowledgements
The Authors thank the members of the Kotake Laboratory for their technical assistance and helpful discussions. This work was supported by JSPS KAKENHI grant number 26430127 (to YK) and the Takeda Science Foundation (to YK).
- Received May 8, 2015.
- Revision received June 29, 2015.
- Accepted July 1, 2015.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Histone H1.2 Represses the Transcription of the p16 Tumor Suppressor Gene
- OIP5-AS1 Promotes Proliferation of Non-small-cell Lung Cancer and Head and Neck Squamous Cell Carcinoma Cells
- Long Noncoding RNA ANROC on the INK4 Locus Functions to Suppress Cell Proliferation
- Long Noncoding RNA, ANRIL, Regulates the Proliferation of Head and Neck Squamous Cell Carcinoma
- The Long Noncoding RNA OIP5-AS1 Is Involved in the Regulation of Cell Proliferation
- Long Noncoding RNA PANDA Positively Regulates Proliferation of Osteosarcoma Cells
- Long Non-coding RNA, PANDA, Contributes to the Stabilization of p53 Tumor Suppressor Protein