


Abstract
Background: The functional contribution of chemokine receptor CXCR7 to malignant brain tumor biology remains controversial. Materials and Methods: Complementary methods were used to confirm CXCR7 expression in clinical glioblastoma multiforme (GBM) specimens and multiple GBM cell lines. Loss-of-function studies were performed using small interfering RNA (siRNA) technology. Results: Elevated CXCR7 levels correlated with reduced survival in glioma patients. CXCR7 was expressed by GBM cell lines and stem-like progenitor cells. Knockdown of CXCR7 by siRNA attenuated phosphorylation of the extracellular signal-regulated kinase (ERK1/2) signaling pathway in response to CXCL12 and resulted in significantly reduced cell proliferation, invasion and migration. Similarly, treatment of glioma cells with a small molecule antagonist of CXCR7, CCX771, significantly inhibited cell proliferation and invasion. Conclusion: CXCR7 actively promotes the proliferation and invasive behavior of glioma tumor cells and stem-like progenitor cells and may be a potential target for glioma therapy.
Chemokines and their receptors play critical roles in many physiological and pathological processes, including cancer, and exert their effects through multiple mechanisms. Chemokine CXCL12 (stromal cell-derived factor 1α, SDF-1α) and its receptor, chemokine (C-X-C motif) receptor 4 (CXCR4), contribute to the growth of primary tumors, as well as progression to metastatic disease. Recently, CXCR7 was identified as second receptor for CXCL12 (1, 2). Elevated levels of CXCR7 correlate with tumor aggressiveness and grade in prostate cancer, metastatic recurrence in non-small cell lung cancer (3, 4), metastasis and mortality of bladder cancer (5), breast cancer (6), pancreatic cancer (7) and survival in cervical cancer (8) and breast cancer (6). In mouse models, CXCR7 enhanced the growth of breast, lung and prostate tumors and experimental lung metastases (3, 6, 9-11). Conversely, tumor growth was dramatically suppressed in prostate cancer (3) and breast cancer (6, 9) xenografts in which CXCR7 expression was knocked-down by siRNA, while direct “nanobody” targeting of CXCR7 reduced head and neck cancer growth in vivo (12).
In vitro mechanistic studies have demonstrated that CXCR7 can facilitate tumor cell resistance to apoptosis and increase cell adhesion (2, 3, 13) and that it plays an essential role in the CXCL12-mediated transendothelial migration of human tumor cells (14). The combined data suggest that CXCR7 can regulate proliferation, angiogenesis, cell trafficking and metastasis in cancer cells.
We originally identified CXCR7 (then called RDC1) as a selective marker of glioblastoma multiforme (GBM) microvascular endothelial cells and demonstrated it was expressed in primary and metastatic brain tumor endothelium, as well as a subset of tumor cells themselves, with little to no expression in corresponding normal cortex (15, 16). Expression of CXCR7 by human gliomas has since been noted by additional groups (17-21) and shown to correlate with tumor grade (17-19, 22). However, the functional contribution of CXCR7 to malignant brain tumor biology remains controversial. For example, Hattermann et al. found that CXCR7 mediated resistance to apoptosis in GBM cells (17, 23) but Liu et al. failed to confirm an anti-apoptotic effect. Instead, they found a high level of heterogeneity in both the surface expression and function of CXCR7, as well as CXCR4 in human primary GBM cells (20).
In the present study, we sought to examine the role of CXCL12/CXCR7 signaling in glioma growth and invasion. We observed that expression of CXCR7 increased in human glioma tissues and that elevated CXCR7 levels were associated with poor survival in clinical glioma patients. Furthermore, we found that CXCR7 was highly expressed in human GBM specimens, as well as multiple GBM cell lines, including U251MG and U373MG cells, and GBM stem-like progenitor cells. Genetic silencing by small interfering RNA (siRNA) or pharmacological inhibition of CXCR7 on these cells impaired glioma cell proliferation and invasion suggesting CXCR7 might be a novel therapeutic target for brain tumor therapy.
Materials and Methods
Meta-analysis of microarray data in human gliomas. Two public microarray databases, Oncomine Research (https://www.oncomine.org) and the Repository for Molecular Brain Neoplasia Data (REMBRANDT) (http://caintegrator-info.nci.nih.gov/rembrandt), were used to study CXCR7 expression in human gliomas. Data of expression of CXCR7 in human gliomas and normal non-neoplastic brain tissues were collected from eight independent studies in Oncomine Research. The REMBRANDT database was used to evaluate the correlation between CXCR7 expression and survival in glioma patients, using a 4-fold expression change for screening as we have in a previous gene expression profiling study (15). Survival plots and statistical significance were generated using the public database software (http://caintegrator-info.nci.nih.gov/rembrandt).
Clinical specimens. Clinical specimens, including GBM and metastatic brain tumor tissues (from lung adenocarcinoma primary), have been described (16). All samples were de-identified according to Health Insurance Portability and Accountability regulations and all study protocols were approved by the IRB of the University of Rochester.
Reverse transcription polymerase chain reaction PCR (RT-PCR). RNA isolation, reverse transcription, PCR preparation was performed as previously described (16).
Cells and reagents. GBM tumor cells U251MG and U373MG cells were gifts of Dr. John Laterra (Johns Hopkins School of Medicine), originally purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), and maintained as previously described (24, 25). GBM stem-like progenitor cells 0913 and SID375 were gifts of Dr. Angelo Vescovi (University of Milan Bicocca, Italy) and Dr. Steven Goldman (University of Rochester, USA), respectively, and maintained as previously reported (26, 27). Basic fibroblast growth factor (FGF) was from Sigma (St. Lous, MO, USA) and used as 20 ng/ml, epidermal growth factor (EGF) was from Invitrogen (Grand Island, NY, USA) and used as 20 ng/ml. Platelet-derived growth factor-AA (PDGF-AA) was from R&D Systems (Minneapolis, MN, USA) and used as 10 ng/ml. Mouse monoclonal anti-human CXCR7 (clone 11G8) for immunohistochemistry was from R & D Systems. Primary CXCR7 antibody for immunoblot analysis was from Abcam (catalogue number: ab12870; Cambridge, MA, USA). Human CXCR7 phycoerythrin antibody and isotype control for flow analysis were from R&D Systems (catalogue numbers: FAB42271P and IC003P, respectively). Primary phospho-p44/p42 MAPK (Erk1/2) and p42/p44 (Erk1/2) antibodies were from Cell Signaling (catalogue numbers: 9106 and 4695, respectively, Beverly, MA, USA). Recombinant human SDF1α/CXCL12 was from Peprotech (Rocky Hill, NJ, USA).
Immunohistochemistry. Paraffin-embedded human GBM sections were obtained from the Department of Pathology, University of Rochester, NY. Immunohistochemsitry was performed as previously described (16). Mouse monoclonal anti-human CXCR7 (clone 11G8) was used at a concentration of 1:500.
Small interfering RNA (siRNA) knockdown of CXCR7. Knockdown of CXCR7 was performed using predesigned CXCR7 Stealth Select siRNA from Invitrogen. The targeted CXCR7-siRNA sequences were GGCUAUGACACGCACUGCUACAUCU (sense) and AGAUGUAGCAGUGCGUGUCAUAGCC (antisense). The negative control CXCR7-scramble sequence has been described (24). Lack of sequence homology between CXCR7, CXCR4 and RefSeq genomes was confirmed using BLAST (NIH) to ensure that all siRNA-CXCR7 constructs were specifically targeted to CXCR7. siRNA was transfected using Lipofectamine RNAiMAX (Invitrogen). Briefly, U251MG and U373MG cells were prepared at 4×105/well in a 6-well plate (BD Bioscience, Franklin Lakes, NJ, USA) and transfected 24 hours later according to manufacturer's protocol. Cells were harvested 48 hours post-transfection and subjected to functional assays. siRNA-scramble and siRNA-CXCR7-transfected lines were designated scramble cells (SC) and siRNA cells (siRNA), respectively.
RNA isolation and reverse transcription. Total RNA was isolated using the Qiagen RNeasy Mini kit (Qiagen, Valencia, CA, USA) according to manufacturer's protocol. cDNA was generated using the iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA, USA) according to manufacturer's protocol.
Quantitative real-time PCR (qPCR). qPCR was performed as previously described (28) by using the iQ5 real-time PCR detection system (Bio-Rad). CXCR7, CXCR4 and GAPDH primers were previously reported (16, 25). All primers were synthesized by Integrated DNA Technologies (IDT, Coralville, IA, USA). Samples were analyzed in triplicates.
Western blotting. To measure CXCR7 protein in U251MG and U373MG cells, cells treated with CXCR7 siRNA for 48 hours were collected and lysed as previously reported (25). Protein concentration was determined by Coomassie protein assay (Pierce, Rockford, IL, USA). Forty micrograms of total protein was loaded on a Novex 10% Tris-Glycine gel (Life Technologies), electrophoresed and transferred to a polyvinylidene difluoride membrane. Blots were incubated with rabbit anti-CXCR7 (1:2000) at 4°C overnight followed by incubation with goat anti-rabbit-horseradish peroxidase secondary antibody (1:5000) for 1 hour at room temperature and visualized with ECL Western Blotting Detection Reagents (Amersham Biosciences, Little Chalfont Buckinghamshire, United Kingdom). To control for protein loading, blots were probed with mouse anti-GAPDH (1:2500, Calbiochem, San Diego, CA, USA) and 1:10,000 rabbit anti-mouse secondary antibody.
To detect the Erk1/2 signal pathway activated by CXCL12, U251MG or U373MG, cells were serum-starved in 10% complete medium for 24 hours then stimulated with CXCL12 (200 ng/ml). Cells were collected at indicated times. Protein was extracted and quantified as described above. Twenty micrograms of protein was loaded on a 4-20% Tris-Glycine gel (Life Technologies). Blots were incubated with primary anti-phospho-p44/p42 MAPK (Erk1/2) antibody (1:1,000) at 4°C overnight followed by incubation with goat anti-mouse-horseradish peroxidase secondary antibody (1:10,000) for 1 hour at room temperature and visualized with Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA, USA). To control for protein loading, blots were stained with rabbit anti-p42/p44 (Erk1/2) antibody (1:1,000) and 1:10,000 goat anti-rabbit secondary antibody.
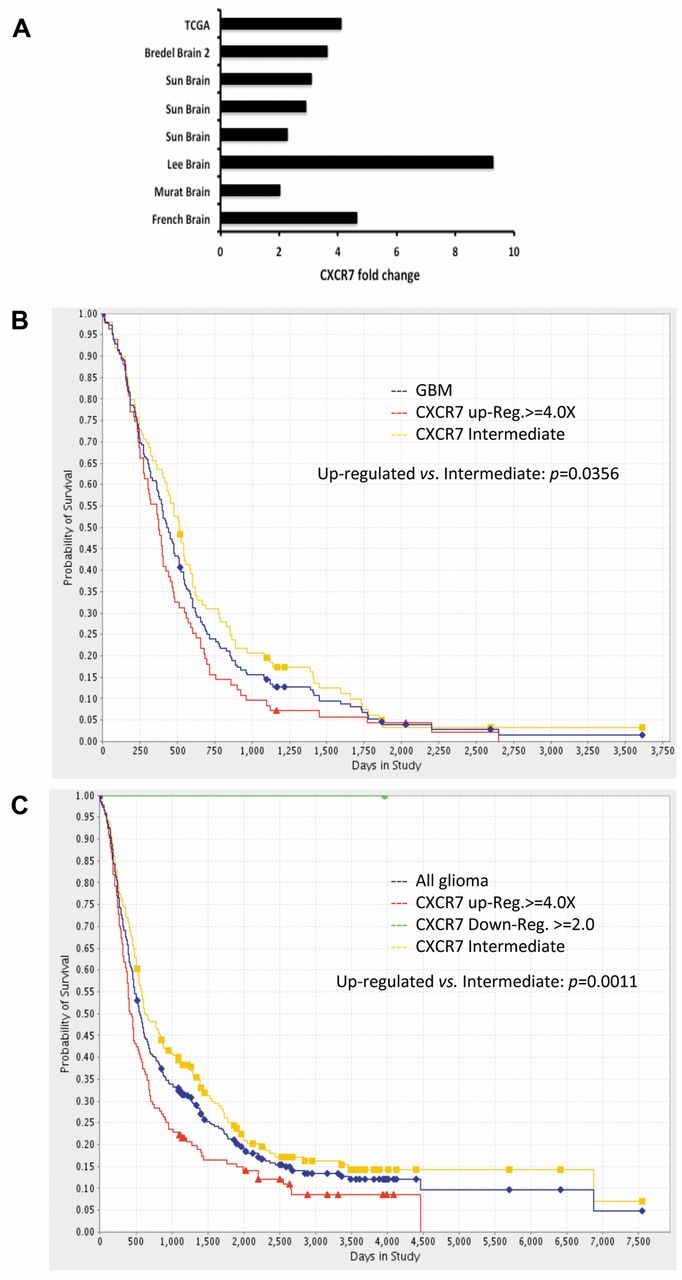
CXCR7 is up-regulated in glioma patients and inversely correlated with patients' survival. A. CXCR7 expression fold changes were collected from eight independent studies in Oncomine Research. Vertical legend is study identification. B and C, Kaplan-Meier plot of data from the Repository for Molecular Brain Neoplasia Data (REMBRANDT) database demonstrates reduced survival amongst GBM patients or glioma patients with 4-fold elevated expression of CXCR7. Survival plots and statistical significance were generated using the public database software.
Flow cytometry analysis. Flow cytometry analysis was performed as described with minor modifications (25). Briefly, glioma cells were collected and suspended in 1% formaldehyde in PBS, incubated on ice for 15 min, followed by addition of 0.2% triton and incubation on ice for another 15 min. Cells were washed with ice-cold washing buffer, then blocked with diluted human FcR blocking reagent (MACS Miltenyi Biotec, San Diego, CA, USA) for 15 min at room temperature. Cells were further incubated with human CXCR7-PE or isotype IgG2A-PE for 30 min on ice, followed by washing with cold washing buffer. Samples were read on a FACSAria IIIU (Becton Dickinson, San Jose, CA, USA) and analyzed with the FLOWJO (10) cytometry analysis software (FLOWJO, Ashland, OR, USA). Ten thousand events were collected per sample.
Cell proliferation assay. Cell proliferation assays were performed using the Cell Proliferation Kit II (Roche Applied Science, Mannheim, Germany) as previously described (25). Briefly, 48 hours after transfection with siRNA, cells were seeded at 1,000 cells/well in 96-well flat bottom plate (BD Bioscience) and cultured in the presence of 200 ng/ml CXCL12. At the indicated time points, 50 μl XTT labeling mixture was added and cells were incubated at 37°C for 4 hours followed by measuring absorbance at 450 nm with a reference wavelength at 620 nm on SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
For CXCR7 blockage experiments, cells were seeded at 1,000 cells/well in a 96-well plate with 200 ng/ml CXCL12 in the presence of 5 μM CCX771 (a specific antagonist of CXCR7) or CCX704 (a negative control analogue with no CXCR7 binding affinity). Both CCX704 and CCX771 were kind gifts of Dr. Mark Penfold, Chemocentryx, Inc. The cell proliferation assay was performed as described above.
Transwell invasion assay. Transwell invasion assays were performed using BD Biocoat growth factor reduced matrigel invasion chambers (25). Invasion chambers were prehydrated with serum-free (0.1% FBS) DMEM (500 μl/well) for 2 hours at 37°C in 5% CO2. After trypsinization, 2.5×104 cells were suspended in 500 μl DMEM (0.1% FBS) and seeded onto the upper chamber, while the lower chamber was filled with 600 μl serum-starved media (0.1% FBS) with 200 ng/ml CXCL12. Cells were allowed to invade for 24 h at 37°C in a 5% CO2 incubator. After 24 h, cells remaining on the upper membrane surface were removed with a Q-tip. The invasive cells on the lower surface of the membrane were fixed and stained with 10 μM Cell Tracker (Invitrogen). Five random 200× fields of adherent cells were counted per well, using a Nikon TE2000-U inverted microscope (Nikon Instruments, Melville, NY, USA) and averaged for statistical analysis.
For CXCR7 blockage experiments, cells were suspended at 2.5×104 cells in 500 μl DMEM (0.1% FBS) with 5 μM CCX771 or CCX704, incubated at 37°C for 30 min and seeded onto the upper chamber, while the lower chamber was filled with 600 μl serum-starved media (0.1% FBS) with CXCL12 (200 ng/ml). After 24 h, invaded cells were counted as described above.
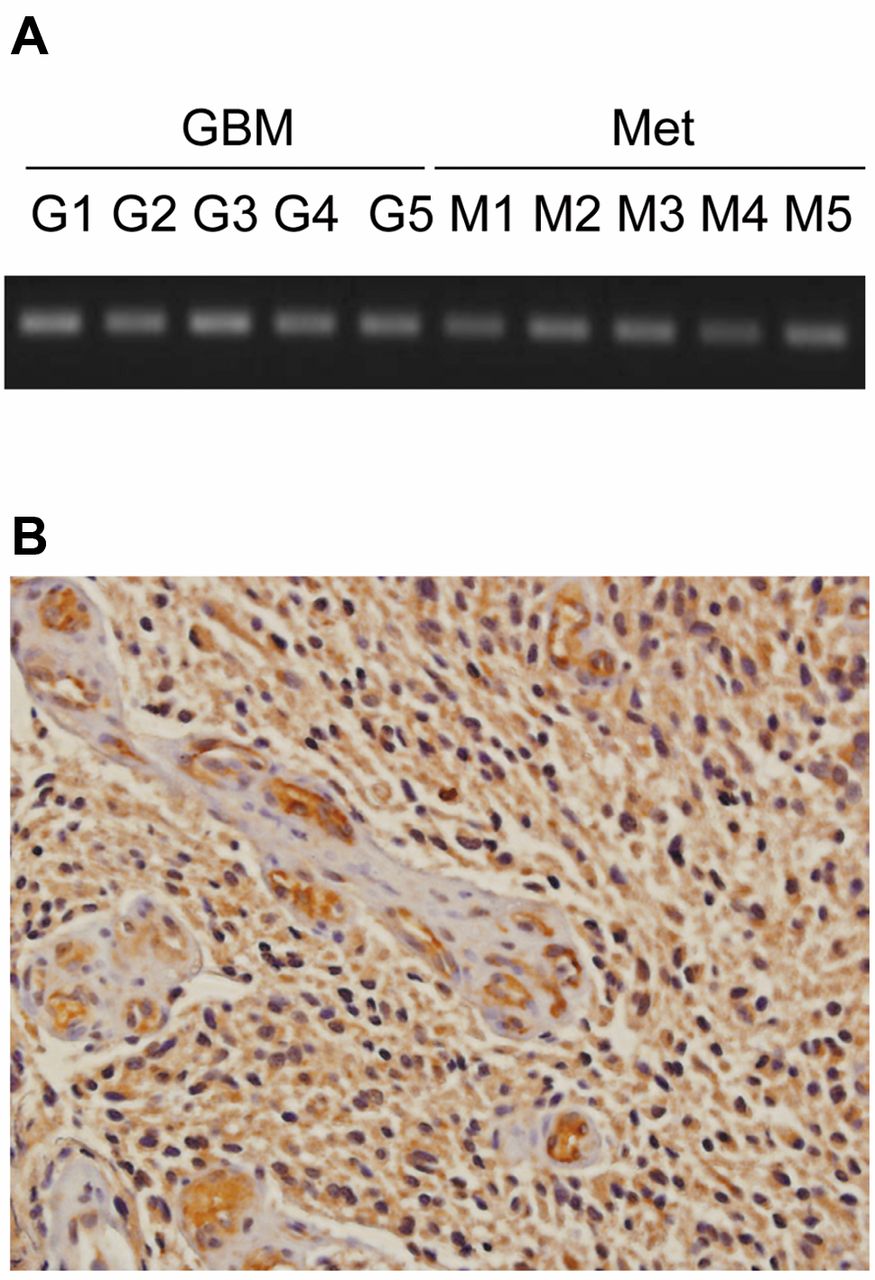
CXCR7 expression in clinical glioma specimens. A. Representative gel pictures for RT-PCR for CXCR7. G, GBM; M, metastatic brain tumor. B. Immunohistochemical staining for CXCR7 in human GBM (magnification: ×200). Paraffin-embedded human GBM sections were obtained from the Department of Pathology University of Rochester, NY. Immunohistochemistry was performed as previously described (16).
Migration assay. Two days after transfection with siRNA, cells were seeded on 6-well plates (6×105/well) and incubated until 100% confluent. After removing media, a 200-μl pipette tip was used to make a scratch across the plastic, denuding the surface of cells. After washing with PBS, cells were cultured in fresh media containing 200 ng/ml CXCL12. At the indicated time, three representative fields at the scratch border were captured using an Axiovert 40 CFL microscope (Zeiss, Thornwood, NY, USA) and PixeLINK software (PixeLINK, Ottawa, ON, Canada). The distance between the two edges of the scratch was measured using the ImageJ software (NIH, http://rsbweb.nih.gov/ij). The distances between the two edges on day 1 and day 2 were normalized to the distance on day 0.
Statistical analysis. Results were analyzed by a two-tailed Student's t-test and differences with p<0.05 were determined as statistically significant. All experiments were repeated three times. Data are shown as mean±standard deviation (SD).

Knockdown of CXCR7 by siRNA. Two days after transfection of glioma cells with CXCR7 siRNA, U251MG cells (A) or U373MG cells (B) were collected and CXCR7 expression was assessed by qPCR (CXCR7; top panels) and western blot (CXCR7; middle panels) as described in Materials and Methods. CXCR4 expression was assessed by qPCR (lower panels) as described in Materials and Methods. For qPCR analysis, three independent experiments were performed. The data represent the mean±SD (**p<0.01, N.S., not significant). For Western blot analysis, three independent experiments were performed and representative data are shown. SC: scramble-siRNA-transfected glioma cells; siRNA: CXCR7-siRNA-transfected glioma cells.

CXCR7 regulates multiple functions in glioma cells. A and B, siRNA-scramble or siRNA-CXCR7-transfected glioma cells were seeded on a 96-well plate and cell proliferation was analyzed as described in Materials and Methods. siRNA-mediated CXCR7 suppression reduced growth of glioma cells. C and D, siRNA-scramble or siRNA-CXCR7-transfected glioma cells were collected and transwell invasion assay was performed as described in Materials and Methods. siRNA-mediated CXCR7 suppression inhibited glioma cells invasion. E and F, siRNA-scramble or siRNA-CXCR7-transfected glioma cells were seeded on a 6-well plate and a migration assay was performed as described in Materials and Methods. siRNA-mediated CXCR7 suppression impeded glioma cells migration. Three independent experiments were performed. All the data represent the mean±SD. SC: Scramble-siRNA-transfected cells; siRNA: CXCR7-siRNA-transfected cells. (*p<0.05; **p<0.01).
Results
CXCR7 increased in human brain tumors and associated with reduced survival. We previously found that CXCR7 mRNA was expressed at levels 9-times higher in malignant brain endothelium than normal brain samples by serial analysis of gene expression (SAGE) and was preferentially localized to vascular regions within glioma samples (15). We expanded these expression data by analyzing microarray data from two public databases: Oncomine Research and the Repository for Molecular Brain Neoplasia Data (REMBRANDT). By examining eight independent studies in Oncomine Research, we found that CXCR7 mRNA significantly increased from 2- to 9-fold in glioma tissues (including GBM, oligodendroglioma and astrocytoma) compared to control non-neoplastic brain tissues. For example, in The Cancer Genome Atlas (TCGA) study, consisting of 542 GBM, CXCR7 mRNA increased 4.112-fold compared to normal brain tissues (Figure 1A). Furthermore, when we analyzed CXCR7 expression in the REMBRANDT database, we found that GBM patients whose tumors demonstrated a 4-fold or greater increase in CXCR7 expression (n=83) had poorer survival than GBM patients without demonstrable CXCR7 elevation (n=99, p=0.0356) (Figure 1B). In addition, when the combined cohorts of low- and high-grade glioma cases were queried, patients with elevated CXCR7 expression (n=144) had a significantly reduced survival compared to patients whose tumors did not exhibit CXCR7 elevation (n=199, p=0.0011) (Figure 1C).

CXCR7-specific antagonist CCX771 inhibited glioma cell proliferation and invasion. A and B, U251MG and U373MG cells were cultured with CCX771 (5 μM) or control CCX704 (5 μM) and cell proliferation assay was performed at indicated time as described in Materials and Methods; C and D, U251MG and U373MG cells were incubated with CCX771 (5 μM) or control CCX704 (5 μM) and transwell invasion assay was performed as described in Materials and Methods. Three independent experiments were performed. All the data represent the mean±SD. (*p<0.05; **p<0.01).
To expand upon the in silico data, we assessed CXCR7 expression in a panel of fresh frozen surgical specimens of GBM and metastatic brain tumor tissues. We found that CXCR7 mRNA was expressed in both primary GBM and metastatic brain tumor specimens (Figure 2A). We confirmed CXCR7 protein expression in human GBM tissue by immunohistochemistry staining using a validated mouse monoclonal antibody (29). Consistent with our previous observation (16), CXCR7 protein was highly expressed in tumor cells, as well as vascular endothelial cells, in surgical specimens of GBM (Figure 2B). Next, we confirmed CXCR7 expression in a variety of glioblastoma cell lines, including U251MG and U373MG cells (Figure 3). By using flow cytometry, we further observed that CXCR7 was expressed both intracellularly and on the cell surface of these glioma lines. We also found that CXCR4 was mainly expressed intracellularly by these same cells with scant cell surface localization (data not shown). With these observations in mind, we sought to investigate the functional role of CXCR7 in glioma cells.
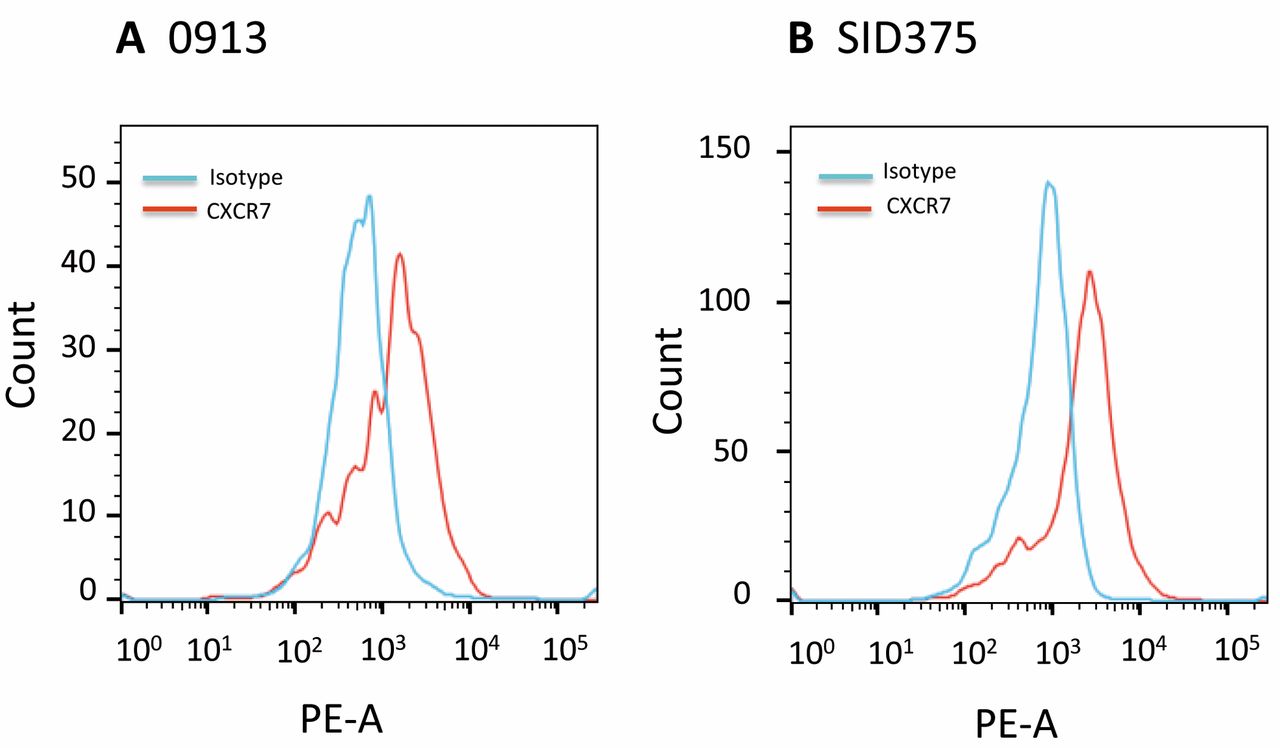
CXCR7 was expressed by GBM stem-like progenitor cells. GBM stem-like progenitor cells (0913 and SID315) were collected and subjected to flow cytometry analysis as described in Materials and Methods. Three independent experiments were performed and representative data are shown.
Suppression of CXCR7 by siRNA inhibits glioma cell proliferation, migration and invasion. To study if CXCR7 is functionally active in glioma cells, we used CXCR7-siRNA to knockdown CXCR7 expression. As shown in Figure 3, we suppressed CXCR7 expression at both mRNA and protein levels in U251MG and U373MG cells using targeted siRNA. Suppression of CXCR7 protein on these cells was also confirmed by flow cytometry (data not shown). To rule-out potential off-target effects of siRNA-CXCR7 on CXCR4, we performed qPCR to check CXCR4 on siRNA-scramble and siRNA-CXCR7 U251MG and U373MG cells and demonstrated that CXCR4 expression remained unchanged (Figure 3, lower panels).
We next used siRNA-CXCR7 and siRNA-scramble glioma cells to study cell proliferation, invasion and migration. We found that inhibition of CXCR7 impeded cell proliferation (Figure 4A and 4B), invasion (Figure 4C, 4D) and migration (Figure 4E, 4F). Cells transfected with CXCR7-siRNA showed statistically significant decreased proliferation, invasion and migration compared to scramble-siRNA-transfected cells (p<0.05 or p<0.01). We similarly confirmed that siRNA mediated knockdown of CXCR7 in U87MG glioma cells inhibited cell proliferation and invasion (data not shown).
A small molecule antagonist of CXCR7, CCX771, inhibits glioma cell proliferation and invasion. While the results using targeted CXCR7 knockdown were encouraging, siRNA remains an impractical approach for glioma management. To develop a more clinically translatable protocol for targeting CXCR7 in glioma, we investigated the effects of CCX771, a small molecule antagonist with high affinity and selectivity for CXCR7 (14). Using the cell proliferation assay and transwell invasion assay, we found that CCX771-treated U251MG and U373MG cells showed significantly decreased proliferation through day 2 to day 4 (p<0.05 or p<0.01; Figure 5A and 5B), as well as a significantly decreased ability to invade through the transwell membrane in response to CXCL12 (p<0.01; Figure 5C and 5D) as compared to negative control CCX704-treated U251MG and U373MG cells.
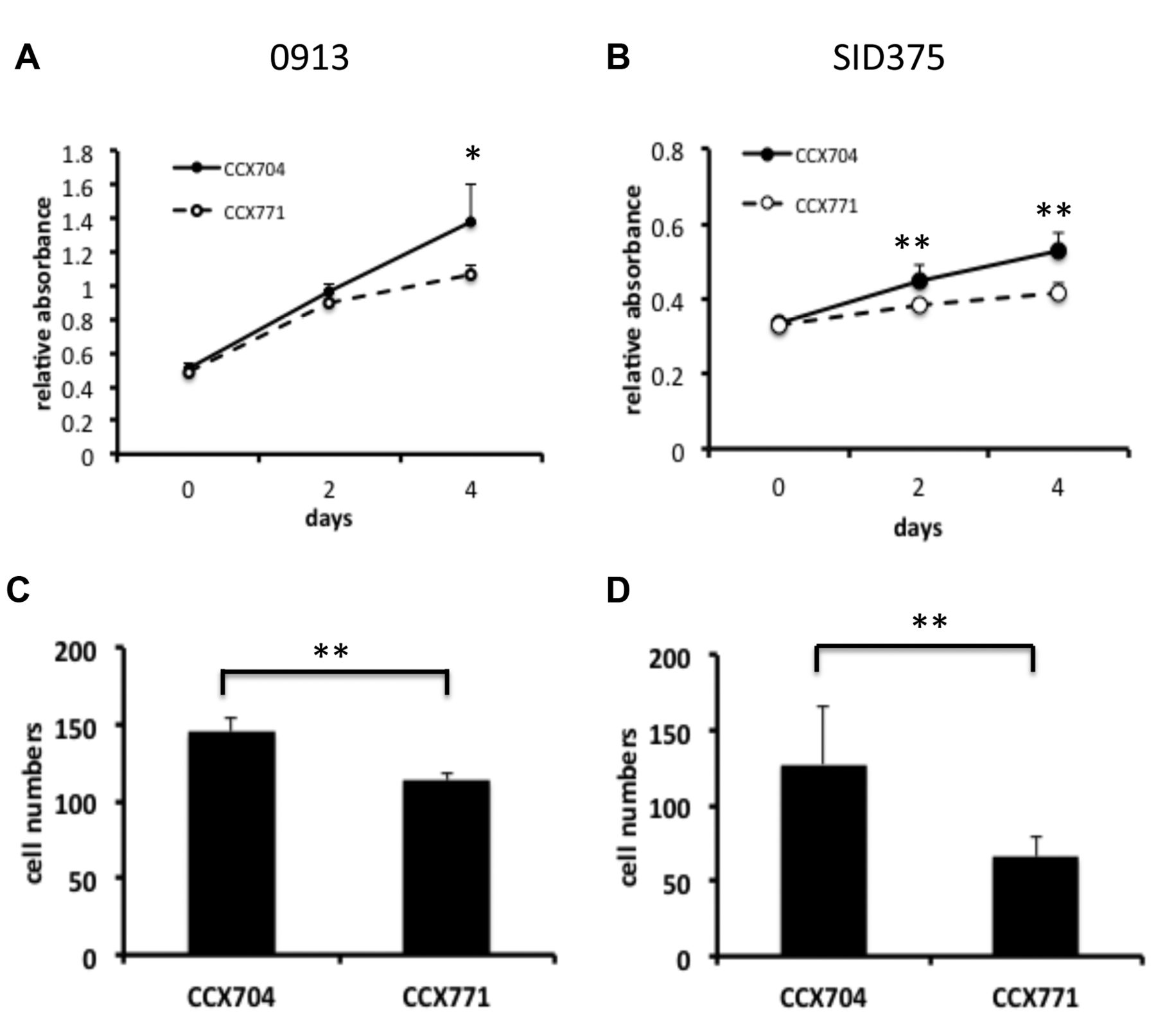
CXCR7-specific antagonist CCX771 inhibited proliferation and invasion of GBM stem-like progenitor cells. A and B, GBM stem-like progenitor cells 0913 and SID375 cells were cultured with CCX771 (5 μM) or control CCX704 (5μM) and cell proliferation assay was performed at indicated time as described in Materials and Methods; C and D, 0913 and SID375 cells were incubated with CCX771 (5 μM) or control CCX704 (5μM) and transwell invasion assay was performed as described in “Materials and Methods”. Three independent experiments were performed. All the data represent the mean±SD. (*p<0.05; **p<0.01).
To further expand our results, we examined the effect of CCX771 on human GBM stem-like progenitor cells. Flow cytometry analysis showed that these cells express CXCR7 (Figure 6). Similar to our observations using traditional GBM cell lines, we found that CCX771-treated GBM stem-like progenitor cells showed significantly decreased proliferation and transwell invasion in response to CXCL12 as compared to CCX704-treated cells (Figure 7). Collectively, these data support our siRNA results and provide a justification for subsequent in vivo targeting of the CXCL12-CXCR7 pathway in glioma xenografts.
CXCR7 knockdown inhibits phosphorylation of ERK1/2. CXCL12 stimulates ERK1/2 phosphorylation in U251MG and U373MG cells. This pathway can be blocked by U0126, a highly selective inhibitor of the upstream ERK kinase, MEK (Liu Y, unpublished data). We treated scramble and siRNA-CXCR7-transfected glioma cells with CXCL12 to assess the impact of CXCR7 expression on the phosphorylation status of ERK1/2. We found that knockdown of CXCR7 in U251MG and U373MG cells by siRNA resulted in 70% and 60% reduction of ERK1/2 phosphorylation in response to CXCL12 (Figure 8), indicating that CXCR7 contributes to CXCL12 mediated activation of ERK1/2 signaling in glioma cells. In contrast, knockdown of CXCR7 had no effect on Akt or p38 phosphorylation (data not shown).

Loss of CXCR7 significantly reduces ERK1/2 phosphorylation in response to CXCL12. Scramble-siRNA or CXCR7-siRNA-transfected glioma cells were serum-starved for 24 hours then stimulated with CXCL12 (200 ng/ml), collected at the indicated times and subjected to western blot as described in Materials and Methods. Three independent experiments were performed and representative data are shown. SC: Scramble-siRNA-transfected glioma cells; siRNA: CXCR7-siRNA-transfected glioma cells.
Discussion
Previous studies have shown that CXCR7 was expressed by human cancer tissues, tumor cell lines and tumor endothelial cells, including clinical brain tumor specimens and glioma cell lines (17-19, 22). However, the functional contribution of CXCR7 to malignant gliomas remains unclear. While Hattermann et al. reported that CXCR7-mediated anti-apoptotic effects (17, 23), Liu et al. failed to observe such a response (20). In order to clarify the role of CXCR7/CXCL12 in malignant brain tumors, we confirmed that CXCR7 mRNA was expressed by both primary GBM and metastatic brain tumor tissues (Figure 2A). The CXCR7 protein was highly expressed in GBM tumor cells, as well as the tumor vasculature (Figure 2B), consistent with our previous observations and as supported by others (16, 17, 19, 22). We found that suppression of CXCR7 by siRNA significantly inhibited proliferation of U251MG, U373MG and U87MG glioma cells (Figure 4A and 4B and data not shown). These data suggest CXCR7 is actively involved in glioma cell proliferation.
GBMs are highly invasive, which directly contributes to the dismal 5-year survival rate for these tumors. In this study, when we examined CXCR7 mRNA expression from eight independent studies from a public microarray database Oncomine Research, we found that CXCR7 mRNA significantly increased from 2- to 9-fold in glioma tissues (including GBM, oligodendroglioma and astrocytoma) compared to control non-neoplastic brain tissues (Figure 1A). Furthermore, by using the REMBRANDT database, we correlated survival data and gene expression microarray data and we found that elevated CXCR7 was associated with poorer survival in both glioma patients in general, as well as GBM patients specifically (Figure 1B and 1C). Therefore, we asked whether CXCR7 could regulate glioma cell migration and invasion. We found that siRNA-mediated suppression of CXCR7 expression in U251MG and U373MG cells significantly impeded cellular invasion and migration (Figures 4C-F) indicating that CXCR7 contributes to glioma cell mobility, as well as proliferation. Targeting CXCR7 in U87MG glioma cells by siRNA produced similar results (data not shown). Conversely, Hatteramn et al. reported that CXCR7 did not alter the migration of U343 or A764 glioma cells in response to CXCL12. This might be due to relatively low dose of CXCL12 used in their study (1 nmol/l) or cell-specific variation (17). However, our data suggest that CXCR7 is functionally involved in glioma cell invasion.
Inhibition of CXCR7 could have direct therapeutic importance for brain tumor patients. CXCR7-specific small molecule antagonist CCX771 has been shown to block CXCR12-medited transendothelial migration of human tumor cells (14). We found that CCX771 significantly impaired the proliferation and invasion of U251MG and U373MG cells in vitro in a manner comparable to siRNA-mediated inhibition (Figure 5). Furthermore, we observed that CCX771 similarly inhibited the proliferation and invasive potential of GBM stem-like progenitor cells (Figure 7). Thus, our findings suggest an active role for CXCR7 in promoting brain tumor growth and the potential feasibility of using CXCR7 antagonists as therapeutic agents for human brain tumors.
CXCL12 may stimulate multiple signaling pathways, depending on the cell type. In the present report, we observed that knockdown of CXCR7 in U251MG and U373MG glioma cells resulted in reduction of ERK1/2 phosphorylation in response to CXCL12 indicating that CXCR7 can regulate ERK1/2 (Figure 8). No reduction in Akt or p38 phosphorylation was detected, suggesting that this was a specific effect and not due to a global dampening of signal transduction. Collectively, our data suggest that CXCR7 is functional in glioma cells and does not solely sequester CXCL12 as reported in other studies (30-33).
In conclusion, CXCR7 actively promotes the proliferation and invasive behavior of glioma tumor cells and stem-like progenitor cells. Together with our recent findings that CXCR7 regulates multiple human brain endothelial cell functions, including proliferation, tube formation and adhesion (25), the collective data suggest that targeting CXCR7 may provide novel opportunities for improving glioma therapy.
Acknowledgements
The Authors would like to thank Dr. Naxin Guo of University of Rochester for technical assistance and advice. They also thank Dr. Su Wang for help with flow cytometry. This work was supported by a University of Rochester Clinical and Translational Science Institute Pilot Studies Award to Y.L., the National Institutes of Health (NINDS K08 NS046461 to K.A.W.), the American Brain Tumor Association (to K.A.W.), the Childhood Brain Tumor Foundation (to K.A.W.) and the Ronald Bittner Brain Tumor Research Fund (to K.A.W.).
Footnotes
-
This article is freely accessible online.
-
Conflicts of Interest
The Authors declare that they have no competing interests.
- Received September 30, 2014.
- Revision received October 21, 2014.
- Accepted October 27, 2014.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}