


Abstract
Background: It remains unclear whether estrogen is produced in prostate cancer (PCa) and how it functions in PCa. Materials and Methods: To examine the production of estrogen in PCa cells, the concentration of estrogen in the medium in which LNCaP cells and PCa-derived stromal cells (PCaSC) were co-cultured, was measured by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS), while aromatase (CYP19) mRNA expression was confirmed by real-time polymerase chain reaction (RT-PCR) methods. To verify whether estrogen is synthesized from testosterone in PCaSC functions, PCaSC were co-cultured with breast cancer MCF-7-E10 cells, which were stably-transfected with ERE-GFP, in the presence of testosterone. GFP expression was detected when PCaSCs could synthesize estrogen. The proliferation of PC-3 cells in the presence of PCaSC was determined by cell count. Results: PCaSC metabolized excessive testosterone to estrogen, which activated estrogen receptor in breast cancer cells. Moreover, estrogen synthesized from testosterone in PCaSC regulated the proliferation of PC-3 cell via repression of some unknown growth factors that were secreted from PCaSC. Conclusion: A chimeric co-culture method between breast cancer cells and PCaSC revealed the production of active estrogen in PCaSC. High-dose testosterone therapy might introduce a new potential strategy to treat CRPC.
- Castration-resistant prostate cancer
- estrogen
- testosterone
- prostate cancer-derived stromal cell
- chimeric coculture
Castration-naïve prostate cancer (PCa) develops into castration-resistant prostate cancer (CRPC) during androgen deprivation therapy (ADT) by various mechanisms. Especially, the androgen receptor (AR) signaling axis plays a key role in this specific development. PCa cells mainly adapt themselves to the environment of lower androgen concentrations and change into androgen-hypersensitive cells or androgen-independent cells. Androgens of adrenal origin and their metabolites synthesized in the microenvironment in an intracrine/paracrine fashion act on surviving PCa cells and secrete prostate specific antigen (PSA). Therefore, new recently developed medicines, abiraterone acetate that inhibits adrenal androgen synthesis enzyme (CYP17A1) and 2nd generation anti-androgen, enzalutamide that not only blocks androgen-AR interaction but also inhibits nuclear translocation of AR and DNA-binding, are clinically effective even after docetaxel-based chemotherapy (1, 2). However, CRPC further develops into a more malignant state after these treatments and finally causes death of patients. One of the mechanisms is that the AR splice variant in which the ligand-binding domain is deleted is constitutively activated without androgen promoting androgen depletion-resistant growth (3). Expression of AR splice variants in PCa bone metastases was associated with castration-resistance and short survival (4). Other mechanisms of development into more malignant CRPC is that CRPC also develops by clonal outgrowth of a small number of androgen-independent PCa cells that preexist or develop at a low frequency due to secondary genetic mutations (5).
Microenvironment, including stromal cells or cancer-associated fibroblasts, also affects PCa cell proliferation, invasion and metastatic progression (6-9). The activation of PCa cell growth through growth factor receptor expression resulted in the activity of androgen-independent stromal growth factor signals, such as fibroblast growth factor (FGF)-7 under conditions of androgen ablation (10). Such a microenvironment may gradually cause clonal development from small number of androgen-independent PCa cells. Therefore, targeting the microenvironment surrounding PCa cells might become a new therapeutic strategy.
Estrogen-based therapies are also alternative ways for CRPC. Excess estrogen causes repression of luteinizing hormone-releasing hormone (LH-RH) secretion from the hypothalamus and subsequently blocks testosterone secretion from testes via repression of LH secretion from the pituitary, as well as LH-RH agonist. Therefore, estrogen-based therapies have been employed as initial hormonal therapies. Estrogen-based therapies are effective not only for castration-naïve PCa as initial treatment but also effective for CRPC (11, 12). However, the molecular mechanisms of effectiveness for CRPC are not fully elucidated yet. Moreover, excess testosterone treatment is effective for CRPC (13, 14). However, it remains unclear why excess testosterone treatment inhibited CRPC growth.
In the present study, stromal cells that co-exist in the microenvironment of PCa cells metabolized excessive testosterone to estrogen and repressed some unknown growth factors, which stimulated PCa cell proliferation.
Materials and Methods
Cell lines. LNCaP cells (ATCC, Manassas, VA, USA) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% penicillin/streptomycin (P/S; Invitrogen, Carlsbad, CA, USA) and 5% fetal bovine serum (FBS; Sigma–Aldrich, St. Louis, MO, USA). PC-3 cells (ATCC) were cultured in RPMI1640 supplemented with 1% P/S (Invitrogen) and 5% FBS. MCF-7-E10 cells were cultured in RPMI1640-10% FCS with 1% P/S. PCaSC which were previously established in our Institution, were cultured in RPMI1640-10% FBS (15).
Cell proliferation assay. Twenty-four hours after plating PC-3 cells at a density of 5×104 onto 12-well plates with DMEM-5% charcoal-stripped fetal calf serum (CCS; Thermo Scientific HyClone, City, UK), cells were cocultured with or without PCaSC-8 onto one layer. Both cells were treated with testosterone (T) or 17β- estradiol (E2) with or without 1 μM bicalutamide or 100 nM exemestane. The media were changed every two days and reagents were also added to the medium. In each experiment, cells were harvested and counted in triplicate using a hemocytometer. The data shown represent the means ± standard deviation (SD) of three replicates.
Real-time reverse transcriptase polymerase chain reaction (RT-PCR). For RT-PCR, 24 h after plating at a density of 2×105 cells onto 6-well plates with DMEM-5% CCS, cells were harvested and total RNA was extracted. Total RNA was purified with the RNeasy mini kit (Qiagen, Valencia, CA, USA). cDNA was made by reverse transcription (RT) of 500 ng of each total RNA using the ThermoScript RT-PCR system (Invitrogen, Carlsbad, CA, USA). One μl aliquot was used as a template for real-time PCR using the CFX connect Real-time PCR system (Bio-Rad, Hercules, CA, USA) according to the manufacturer's instructions. PCR for AR, aromatase (CYP19), estrogen receptor α (ERα), ERβ and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was performed according to the manufacturer's instructions (Bio-Rad). The primers were AR sense: 5’-TCCAAATCACCCCCCAGGAA-3’ and antisense: 5’-GACATCTGAAAGGGGGCATG-3’; aromatase (CYP19) sense: 5’-GGAACACTAGAGAAGGCTGGTCAGT-3’ and antisense: 5’-GCCTCGGGTCTTTATGGATACGGTT-3’; ERα sense: 5’-CACTGCGGGCTCTACTTCATCGCA-3’ and antisense: 5’AAGAGCTACGGGAATCCTCACGCTT-3’; ERβ sense: 5’-CGGCTTTGTGGAGCTCAGCCTGTTC-3’ and antisense: 5’-GCCGCTCTTGGCAATCACCCAAACC-3’; GAPDH sense: 5’-CCACCCATGGCAAATTCCATGGCA-3’ and antisense: 5’-TCTAGACGGCAGGTCAGGTCCACC-3’. The annealing temperature of RT-PCR was 56°C for AR, and 65°C for aromatase (CYP19), ERα, ERβ and GAPDH.

DHT and E2 biosynthesis from androgens in LNCaP cells and PCaSC-8. The concentration of T and E2 in the medium used for co-culture with LNCaP and PCaSC after treating with 100 nM DHEA, 10 nM AD, Adiol, T and DHT is shown. Twelve hours after starting culture of 5×104 LNCaP cells with or without 5×104 PCaSC-8, 100 nM DHEA, 10 nM AD, Adiol, T or DHT were added to the medium. Then, aliquots of medium were collected after 24 h for measuring the concentration of DHT and E2 by LC–MS/MS. The lower limit of quantitation of DHT and E2 was 2.5 and 0.03 pg/assay, respectively.
Detection of estrogen receptor (ER)-activating ability of PCaSCs. ER-activating abilities of PCaSCs were detected by coculture with MCF-7-E10 cells, ER activity reporter cells, as described in a previous study (16). MCF-7-E10 cells were established from human breast cancer MCF-7 cells by stable transfection with the estrogen response element (ERE)-green fluorescent protein (GFP) (ERE-GFP) reporter plasmid. These cells express GFP in the presence of estrogen. In the coculture of prostate cancer-CAFs (PCaSCs) and MCF-7-E10 cells, they were precultured in estrogen-deprived medium for 3 days. PCaSCs were seeded in a 24-well multi-well dish at 5×104 /ml, and on the next day the same number of MCF-7-E10 cells was seeded on the top of PCaSCs. Subsequently, the cellular mix was cultured for 4 days in the presence or absence of testosterone (100 nM) and/or fulvestrant (ICI 182780; 1 μM) or exemestane (100 nM). MCF-7-E10 cells expressing GFP were counted after collected with trypsinization. PCaSCs and MCF-7-E10 cells were easily discriminated by their morphology. Breast cancer-CAFs (#863, 870, 871,872 and 874) were used as a positive control.

Quantification of AR, aromatase (CYP19), ERα and ERβ mRNA expression level in LNCaP, PC-3, PCaSC-5 and PCaSC-8. The expression level of each PCR product was quantitated and normalized by the GAPDH expression level. The relative level of expression in each cell line was adjusted to the level of expression of LNCaP cells.
Liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS). After plating 5×104 LNCaP cells on 12-well plates in DMEM-5% CCS, LNCaP cells were co-cultured with PCaSC-8 for 24 h. Both cell lines were treated with 100 nM dehydroepiandrosterone (DHEA), 10 nM androstenedione (AD), 10 nM androstenediol (Adiol), 10 nM testosterone (T) or 10 nM dihydrotestosterone (DHT) for 24 h and then the media were collected. The concentration of estradiol (E2) in the media was measured by LC-MS/MS (Division of Pharmacological Research, Aska Pharma Medical Co. Ltd., Kawasaki, Japan).
Results
Synthesis of estrogen in PCa-derived stromal cells. We have previously shown that PCa-derived stromal cells (PCaSCs) expressed androgen synthesis enzymes and had the ability to synthesize testosterone and DHT from DHEA in PCa tissue (15). We here confirmed how DHEA, AD, -Adiol, T, DHT are metabolized to DHT and β-estradiol in the presence or absence of stromal cells. PCaSC-8 increased the concentration of testosterone and DHT when DHEA was added as substrate to the medium of LNCaP cells (Figure 1). Interestingly, PCaSC-8 reduced the concentration of testosterone and DHT when androstenediol, testosterone and DHT were added as substrates to the medium of LNCaP cells. Next we examined the expression of AR, ERα, ERβ, aromatase (CYP19) mRNA in LNCaP, PCaSC-5 and PCaSC-8 cells. Although ERα mRNA in well-expressed in all cells, ERβ mRNA was expressed in PC-3 cells only. The expression of the aromatase (CYP19) mRNA in PCaSC-5 and PCaSC-8 was relatively high compared with LNCaP and PC-3 cells (Figure 2). We also examined the concentration of estradiol (E2) in the medium of LNCaP cells co-cultured with PCaSC-8. Production of E2 was observed in LNCaP cells in the presence of 10 nM testosterone (Figure 1). Moreover, coculture with PCaSC-8 elevated the production of E2 in LNCaP cells treated with 100 nM DHEA, 10 nM AD, 10 nM Adiol and 10 nM T suggesting that PCaSC-8 had a strong ability to synthesize E2 by aromatase.

Detection of estrogen receptor (ER)-activating ability of PCaSC. After 3 days of culture in PRF-RPMI with 10% DCC-FCS, PCaSC-5 or PCaSC-8 were co-cultured with E10 cells in the presence or absence of 100 nM of T for 4 days with or without 1 μM ICI 182780 or 100 nM exemestane (Exe). Then, the numbers of E10 cells expressing GFP were counted and the data are expressed as percentages of GFP-expressing cells. Breast cancer-CAFs (#863, 870, 871,872 and 874) were used as a positive control. The data are presented as the mean±SD of triplicate measurements.
Activity of E2 from PCaSCs. Although PCaSCs secreted E2 to the medium, it remains uncertain whether the secreted E2 from PCaSCs has physiological activity. To examine the activity of E2, we employed the unique approach by Yamaguchi et al., who established a new reporter cell system (16). To visualize the activation of estrogen receptor (ER) by estrogen, they established a stable transformant-named E10-of human breast cancer MCF-7 cells, by transfection with the estrogen-responsive element-green fluorescent protein (GFP) gene. E10 cells specifically express GFP when ER is activated by estrogen or by co-culture with stromal cells isolated from breast tumor tissues in the presence of testosterone, a substrate for aromatase. Therefore, we used PCaSCs and testosterone instead of stromal cells from breast tumor tissues to investigate the activation of ER by PCaSCs (Figure 3). One nM E2 increased the number of GFP-positive E10 cells in the absence of stromal cells and this effect was inhibited by anti-estrogen, 1 nM ICI 182780 and aromatase inhibitor, exemestane (Exe). One-hundred nM of testosterone did not increase the number of GFP-positive E10 cells. Breast cancer-derived stromal cells increased the number of GFP-positive E10 cells in the presence of 100 nM testosterone. Next, we co-cultured E10 cells with PCaSC-5 or PCaSC-8 in the presence of testosterone. One hundred nM of testosterone increased the number of GFP-positive E10 cells in the presence of PCaSC-5 or PCaSC-8 and this increase was reduced by ICI 182780 and Exe suggesting that PCaSCs have the ability to synthesize estrogen and secrete active estrogen to the medium.
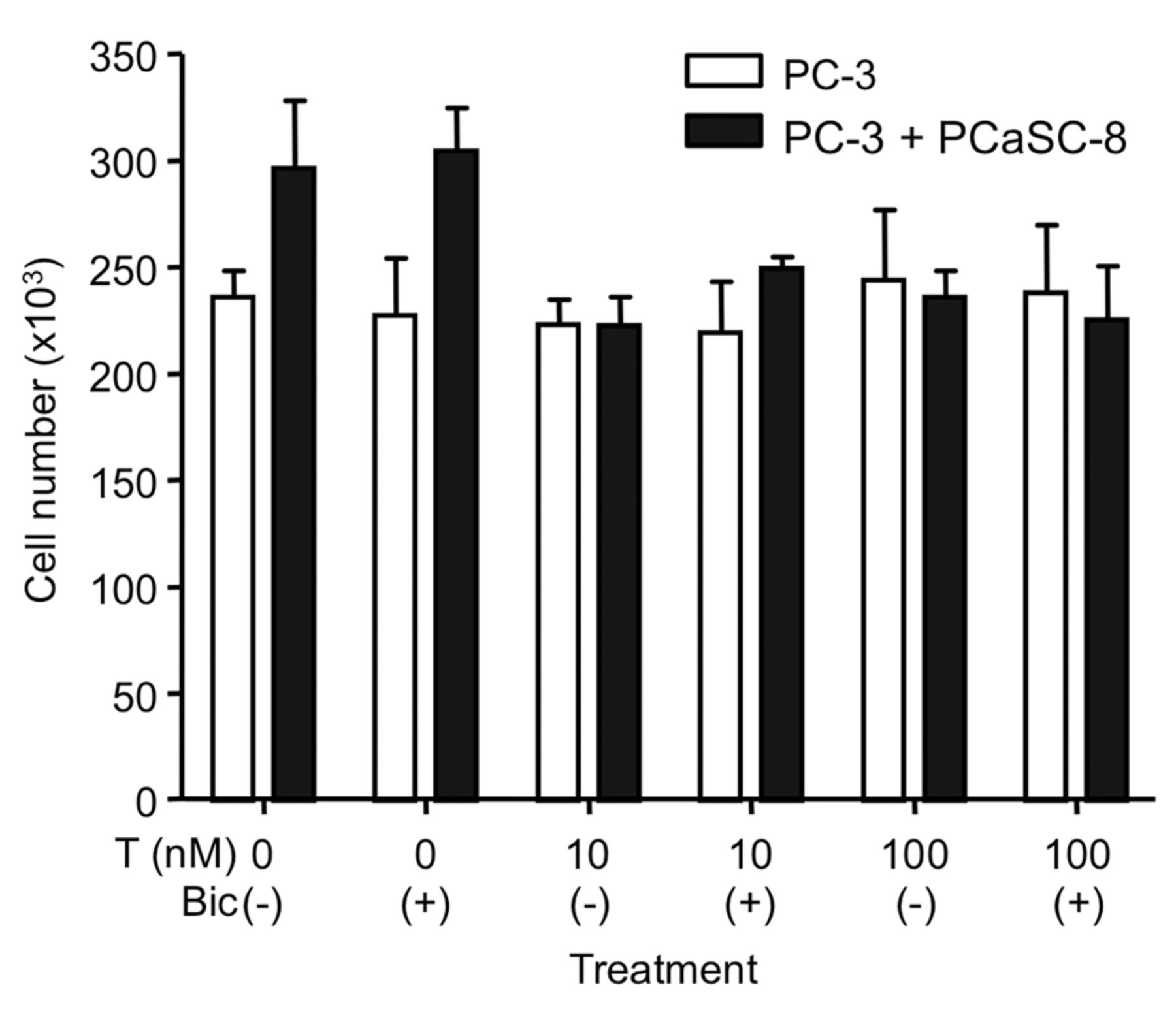
Effect of PCaSC-8 and T on PC-3 cell proliferation. Twenty-four hours after 1×104 PC-3 cells were cultured on the lower chamber, PCaSC-8 were seeded on the upper chamber. Twelve hours later, cells were treated with T with or without 1 μM bicalutamide for 4 days and counted. The medium was changed every 2 days and T was added to medium. These coculture experiments were performed at least twice with reproducible data. The data are presented as the mean±SD of triplicate measurements.
Inhibition by testosterone of PC-3 cell proliferation is accelerated by PCaSC. Since PC-3 cells do not express AR, testosterone could not affect the proliferation of PC-3 cells (Figure 4). Thus, when androgen-independent PC-3 cells were cocultured with PCaSC-8, the proliferation of PC-3 cells was stimulated by PCaSC-8. This effect was blocked by the addition of 10 or 100 nM of testosterone (Figure 4). To confirm whether the effect of testosterone was mediated through the AR in PCaSC-8, we treated PC-3 cells that were cocultured with PCaSC-8 with 10 μM antiandrogen, bicalutamide, in the presence of testosterone. However, bicalutamide could not recover the inhibition by testosterone. Then, 0.1 or 1 nM of estradiol was added to PC-3 cells that had been cocultured with PCaSC-8. Estradiol inhibited the proliferation of PC-3 cells, initially stimulated by PCaSC-8 (Figure 5). Moreover, the inhibition by testosterone in the presence of PCaSC-8 was also blocked by the aromatase inhibitor exemestane, suggesting that inhibition by testosterone is mediated through ER after conversion of testosterone to estradiol in PCaSC-8.
Discussion
In the present study, we revealed that PCaSC have the ability to synthesize estradiol from testosterone. Moreover, PCaSC secrete cytokine(s), which stimulate PCa cells. The function of cytokine(s) was regulated by estradiol and estradiol synthesized from testosterone.
The physiological function of estradiol synthesized in stromal cells remains unclear. Physiologically increased estrogens by aromatase over-expression in mouse promoted the development of prostatitis and prostatic intraepithelial neoplasia lesions (17). Estradiol exerted a synergistic effect with androgens in inducing glandular prostatic hyperplasia in castrated dogs (18). Concentrations of estradiol as low as 0.001 nM were sufficient to significantly increase growth rates in normal prostate stromal cells, whereas the benign prostatic hyperplasia (BPH) stromal cells behaved similarly to the normal stromal cells at concentrations of 0.1 nM and above (19). Moreover, the addition of E2 was able to increase the levels of total transforming growth factor (TGF)-β1 in the BPH-derived stromal cells (19). Exogenous estradiol (10−7 and 10−6 M) moderately increased the proliferation of stromal cells in culture. However, estradiol had no effect on the proliferation of epithelial cells in culture (20).
In contrast, the pharmacological effect of estrogen on PCa is totally different. High-dose estrogen therapy using synthetic estrogen, diethylstilbestrol (DES), for androgen-naïve PCa has been initiated since 1941 when Huggins et al. proposed the concept of hormonal therapy (21). This effect is mediated through a negative feedback regulation on the hypothalamic-hypophyseal-testicular pathway. DES and ethinylestradiol are eventually useful agents to treat not only androgen-naïve PCa but also CRPC (12, 22, 23). It remains unclear, however, why exogenous estrogen is effective for CRPC. Exogenous estradiol suppressed angiogenesis of androgen-responsive PCa (24). Estrogen receptor α in cancer-associated fibroblasts suppressed PCa invasion via modulation of thrombospondin 2 and matrix metalloproteinase 3 (25). Recently, based upon findings in an animal model (13, 26), a phase I trial of high-dose exogenous testosterone in patients with CRPC was conducted and serum PSA level was decreased in 7 out of 12 patients. The authors described that one of the mechanisms of tumor regression is the biphasic effect of testosterone in LNCaP cell growth (27). Stemming from our results, we propose another mechanism suggesting that the effect of high-dose testosterone might be mediated through the metabolism of testosterone to estradiol and suppression of growth factors from stromal cells by estradiol.
In conclusion, chimeric co-cultures between breast cancer cells and PCaSC revealed that production of estrogen in PCaSC caused the repression of growth factors from PCaSC. This result might introduce a new potential strategy to treat CRPC by estrogen or high-dose testosterone. We are currently studying candidate growth factors that are regulated by estradiol in PCaSC since their identification may provide new strategies to cure CRPC.

Effect of PCaSC-8 and T and E2 on PC-3 cell proliferation. Twenty-four hours after 1×104 PC-3 cells were cultured on the lower chamber, PCaSC-8 were seeded on the upper chamber. Twelve hours later, cells were treated with T or E2 with or without 100 nM exemestane (Exe) for 4 days and counted. The medium was changed every 2 days and T was added to medium. These coculture experiments were performed at least twice with reproducible data. The data are presented as the mean±SD of triplicate measurements.
Acknowledgements
The Authors would like to thank appreciate Ms. Fujinuma for technical support. A Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sport, Science, and Technology of Japan (25670679).
Footnotes
-
Conflicts of Interest
The Authors declare no conflict of interest.
- Received October 1, 2014.
- Revision received November 18, 2014.
- Accepted November 19, 2014.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.