


Abstract
Natural-killer group 2, member D (NKG2D) is an activating receptor found on activated natural killer cells and on activated T-cells, here termed cytokine-activated killer (CAK) cells. NKG2D ligands are expressed on various human cancer types. Gemcitabine is an anticancer drug which is a less immune-destructive agent than others. Herein, we investigated the clinical efficacy and the underlying mechanisms of a combination of CAK cell infusion therapy and gemcitabine. Twenty-three patients with disseminated carcinomas were treated with chemo-immunotherapy consisting of CAK cell infusion therapy following gemcitabine treatment. To investigate the underlying mechanisms by which CAK cells synergize with gemcitabine, we used enzyme-linked immunosorbent assay, Real-time reverse transcription polymerase chain reaction assay, calcein-release assay, and adherent target detachment assay. Using these assays we determined the NKG2D ligands such as major histocompatibility complex-class I-related chain (MIC)A/B expression in carcinoma cells and the level of cellular cytotoxicity generated by treatment with gemcitabine with/without CAK cells. The tumor responses differed among the patients (n=23). In vitro experiments revealed that MICA/B protein and mRNA expression were up-regulated in several carcinoma cell lines after gemcitabine treatment. Pre-treatment with gemcitabine and subsequent exposure to CAK cells induced greater cytotoxicity than either treatment alone. Inclusion of soluble MICB in CAK cell-mediated cytotoxicity assay significantly reduced cytotoxicity. Our clinical results of gemcitabine–CAK combinatorial therapy demonstrated long-term stable disease despite chemoresistance. In conclusion, the combination of gemcitabine and CAK cells may have clinical therapeutic significance for pancreatic, hepato-biliary tract, and urothelial tract cancer. Our study shows that combining CAK therapy with gemcitabine can lead to successful treatment of metastatic cancer.
Natural killer (NK) cells, γδ+ T-cells, and activated CD8+ T-cells are important effectors in the immune response to tumors, and have been used in cell transfer therapy for various types of cancer (1-5). A number of inhibitory and activating receptors on the surface of these cells tightly regulate their interaction with target cell ligands (1, 2). In particular, the strength of the antitumor immune response appears to be critically dependent on the surface levels of the activating receptor, natural-killer group 2, member D (NKG2D) (5, 6). Correspondingly, expression of NKG2D ligands on target cells is a requirement for effective tumor immunosurveillance and the elimination of cancer cells. Major histocompatibility complex class I-related chain A and B (MICA and MICB, respectively) are polymorphic transmembrane glycoproteins and are NKG2D ligands. Expression of MICA/B can be induced by various cellular and environmental stimuli, including heat shock, virus infection and DNA damage-inducing agents (7, 8).
Cytokine-activated killer (CAK) cells, as termed here, consist of activated NKG2Dhigh T-cells and activated Natural Killer cells. Both activated T-cells and activated NK cells have been shown to induce cytotoxicity against various types of cancer in a manner independent of T-cell receptor–MHC class I interactions. Previously, we studied the in vitro effect of CAK cells in combination with gemcitabine against hepatocellular carcinoma cells (9). Herein, we investigated the feasibility and potential of CAK therapy in combination with gemcitabine.
Gemcitabine, a potent chemotherapeutic agent against various types of cancers, including cancer of the pancreas, hepato-biliary tract, and urothelial tract, has recently been proposed to have immunomodulatory effects (10, 11). Nowak et al. showed that gemcitabine suppressed IgG antibody production, but did not block lymphocyte recall response and was not detrimental to specific antitumor immunity (12). It has also been found that gemcitabine does not diminish cellular immunology, suggesting that cellular and molecular therapies could, therefore, be used in parallel to improve their effect compared to chemotherapy-alone (11, 12). Suzuki et al. demonstrated that gemcitabine-resistant tumor cells were sensitive to in vivo murine gemcitabine treatment, suggesting a gemcitabine-mediated enhancement of T-cell-mediated antitumor effects (13). Indeed, Bauer et al. have reported that a combination of a dendritic cell vaccine with gemcitabine was effective in a pancreatic cancer model (14). Thus, gemcitabine appears to be a good candidate drug for combination with immunotherapy.
We previously reported that gemcitabine induced MICA/B expression on hepatocellular carcinoma cells and an increase in susceptibility to lysis by CAK cells (9). Because gemcitabine is clinically used against pancreatic, hepatobiliary, urothelial tract, and non-small cell lung cancer, it is interesting to study whether combinatorial use of gemcitabine and CAK therapy is effective.
Chemo-immunotherapy has gained much attention in recent years. Several studies have demonstrated that chemotherapeutic agents such as oxaliplatin can induce immunogenic cell death in various types of cancer cells through releasing calreticulin and high mobility group box 1 (HMGB1) from tumor cells and inducing antigen pickup by dendritic cells (15, 16). Moreover, non-myeloablative chemotherapy has several benefits, such as long-term survival and tumor-site infiltration of activated lymphocytes (17, 18).
In the present study, we show the clinical response to a combination therapy consisting of gemcitabine and CAK therapy using retrospective analysis. We also demonstrate the mechanisms by which CAK therapy synergizes antitumor effects in combination with gemcitabine treatment. Our study warrants further randomized clinical studies of combination therapy of gemcitabine and CAK administration therapy.
Patients and Methods
Patients. Clinical and immunological parameters of 23 patients cancer were studied in this retrospective study. Patients had histologically proven cancer with verified progressive metastatic disease stage IV. Fifteen patients with pancreatic cancer, five with biliary duct cancer, two with non-small cell lung cancer, and one with urinary bladder cancer were included. Written informed consents were obtained from these patients. Disease in all patients was resistant to standard therapies, including surgery, radiation and standard chemotherapy alone. The cell processing and adoptive immunotherapy procedures used were approved by the Ethics Committee of our institution.
Treatment protocol. Patients received gemcitabine every four weeks as two or three consecutive infusions of 800-1,000 mg/m2 in combination with the CAK therapy described below. At various time points before and after treatment, the tumor response was assessed by a tumor marker and imaging (Computed Tomography and Magnetic Reasonance imaging), and according to the Response Evaluation Criteria in Solid Tumors 1.0 (RECIST) (19). In each cycle, patients received gemcitabine therapy at a hospital, and 4-5 days later they received CAK therapy at our clinic. They were given at least 12 therapy cycles and were followed-up for more than 12 months.
Preparation of CAK cells and treatment. Peripheral blood mononuclear cells (PBMCs) were collected with a blood cell separator (Haemonetics CCS, Haemonetics Corporation, Braintree, MA, USA) and cryopreserved at −80°C. PBMCs (2×107-4×107 cells) were divided into two batches and cultured by two different methods for seven days. In one method, the cells were stimulated with both human recombinant interleukin (IL)-2 (rIL2, 200 U/ml; ILCASS, Laboratorio Pablo Cassara S.T.C. Argentina, Buenos Aires, Argentina) and 5 μg/ml antibody to OKT3 (Orthoclone® OKT3, Janssen Pharmaceutical, Tokyo, Japan) for NKG2Dhigh T-cell activation, and in the other method, the cells were stimulated with a high dose of IL2 (2,000 U/ml) alone to increase the number of activated NK cells. In this study, CAK cells are NKG2Dhigh CD8+ T-cells and activated NK cells. After seven days in culture, PBMCs were transferred to a culture bag system (KBM550, Kohjin Bio, Osaka, Japan, or ALys505N, Nipro, Osaka, Japan) and expanded for seven days to obtain sufficient numbers of CAK cells. All cultures were inspected for safety by examining contamination of endotoxin, β-glucan and peptide-glycan with Toxinometer ET-6000 (Wako Pure Chemical Industries, Ltd., Osaka, Japan) following the Food and Drug Administration guidelines. Mycoplasma contamination was inspected by MycoAlert (Lonza Rockland Inc., Rockland, ME, USA). The cell processing and immunotherapy procedures used were approved by the Ethical Committee of our institution.
mAb reagents and flow cytometry. Surface markers on lymphocytes or CAK cells were labeled by direct or indirect immunofluorescence using monoclonal antibodies[CD3-fluorescein isothiocyanate (FITC), CD4-FITC, CD8-PE, CD16-FITC, CD56-Phycoerythrin (PE) (Immunotech Beckman Coulter, Brea, CA, USA); NKG2D-PE (R&D Systems, Minneapolis, MN, USA)]. Cell-surface expression of MICA/B was detected using flow cytometry. Fluorescence was detected using an FC500 flow cytometer (Beckman Coulter) and expressed as relative mean fluorescence intensity (MFI) or percentage above baseline. For tumor cell surface marker expression, detached cells were washed twice in cold Phosphate Buffered Saline (PBS) and stained with PE-labeled mouse antibody to human MICA/B (R&D Systems) for 60 min at 4°C. Anti-human UL16-binding protein (ULBP)-1 and ULBP3, Class I were purchased from R&D Systems.
Culturing tumor cell lines. KLM-1 pancreatic carcinoma cell line and T24 urothelial carcinoma cell line were obtained from RIKEN Cell Bank (Tsukuba, Japan), and were cultured in RPMI medium supplemented with 5% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin/streptomycin (Meiji Seika, Tokyo, Japan). Viable cells were counted using Cell Counting Kit-8 (Dojindo, Osaka, Japan). Cells were treated with different amounts of gemcitabine, or with Dimethylsuphoxide (DMSO) vehicle control. DMSO concentrations were less than 0.1%. Cells were counted with FD500 (Sysmex, Osaka, Japan). For WST-8 proliferation assays, cells were cultured in 96-well dishes (Nunc, Nalgene, Thermo Scientific) in 100 μl at a density of 5×104 cells/ml. Assays were performed over 48 h, using a minimum of five replicates. For co-incubation with drugs, cells were treated with different concentrations of gemcitabine (10 ng/ml-10 μg/ml). Proliferation assays were performed using the WST-8 cell proliferation kit (Dojindo) according to the manufacturer's instructions.
Clinical characteristics of patients.
Cytotoxicity assay. We modified an adherent target detachment assay described previously (20, 21) to measure the cytotoxicity of CAK cells. Target cells (5,000 per well) were seeded in a 96-well flat bottom plate and incubated for 24 h to allow adherence. Gemcitabine (0-10 μg/ml) was then added to wells and cells were incubated for another 24 h prior to addition of effector cells at an effector:target (E:T) cell ratio of 20:1, 40:1, or 80:1. Target and effector cells were then incubated for 4 h. Dead target cells detached from the culture surface and were recovered by washing, together with the added effector cells. To quantify the viable adherent cells, WST-8 reagent solution (from the Cell Counting Kit-8) was added to the washed wells and cells incubated for 1 h at 37°C. The absorbance at 450 nm was then measured using a microplate reader (ImmunoMini NJ-2300; Nalgen, Nunc, Thermo Scientific, Waltham, MA, USA). Detached cells were stained with 7-amino-actinomycin.D (BD Biosciences, San Jose, CA, USA) to confirm that the detached tumor cells were indeed non-viable. In some experiments, soluble recombinant MICB (American Research Products, Inc.™ Belmont, MA, USA) was added at different concentrations to the tumor cells followed by 4 h incubation prior to addition of CAK cells.
Calcein-release cytotoxicity assay and cell imaging. Antibody-dependent cellular cytotoxicity assays were performed using calcein AM release. Briefly, target cells (KLM-1 and T24: Riken Cell Bunk, Tsukuba, Japan) were resuspended at 1×105 cells/ml in complete medium and left to adhere to a plastic culture surface. After overnight culture, cells were incubated at 37°C for 1 h in the presence of calcein AM, before CAK cells at an E:T ratio of 20:1, 40:1, or 80:1 with/without gemcitabine-pretreatment were added to the culture. After 4 h incubation at 37°C, the plate was washed and adherent cells were subjected to analysis under a fluorescence microscope driven by Lumina Vision software (version 2.4.2; Mitani Corp, Fukui, Japan). Images were captured using a conventional fluorescence microscope (IX81; Olympus, Tokyo, Japan) equipped with a color CCD camera (DPI72; Olympus) and objective lens (LUC plan FLN; Olympus). All procedures were performed at 20-25°C. Images were analyzed using Lumina Vision software. During observation, cells were warmed to 37°C on a thermoplate (MATS-U55R30; Tokai Hit, Shizuoka, Japan). The cytotoxicity percentage was calculated as: control fluorescence − sample fluorescence/control fluorescence ×100%.
Lymphocyte surface markers before and after activation.
Quantitative real-time polymerase chain reaction. To perform real-time reverse transcription-PCR, total RNA was isolated using High Pure RNA Isolation kit (Roche Diagnostic GmbH, Mannheim, Germany) followed by DNase digestion and reverse transcription using a Transcriptor High Fidelity cDNA Synthesis kit (Roche Diagnostic GmbH). Primer sequences were as follows: MICB, 5’-ACCTTGCTATGAAGGTCACA-3’ (forward) and 5’-CCCTCT GAGACGTCGCTGCA-3’ (reverse), Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5’-ATGACATCAAGAAGGT GGTG-3’ (forward) and 5’-CATACCAGGAAATYGAGCTTG-3’ (reverse). The resulting cDNA was amplified with GAPDH- and MICB-specific primers (35 cycles: denaturation at 95°C for 10 s, annealing at 55°C for 5 s, and extension at 72°C for 5 s) using SYBR Green with a Light Cycler (Roche). Relative MICB mRNA expression was calculated by normalization against GAPDH expression.
Statistical analysis. All data are expressed as mean±SEM. Differences between groups were assessed for statistical significance using the Mann–Whitney test or paired Student's t-test depending on the distribution of the data; p<0.05 was considered statistically significant.
Results
Patients' characteristics. Patients with metastatic solid tumors that were treated with gemcitabine in combination with adoptive transfer of CAK cells for more than six months are shown. The clinical and immunological characteristics of the patients and their CAK cells are shown in Tables I and II, respectively. All patients had stage IV advanced tumors with visceral or peritoneal metastasis, including the liver, lung, lymph nodes, bone and soft tissue. Prior to therapy, 6 out of 23 patients had been treated with surgery/chemotherapy, and had progressive disease refractory to treatment. All patients completed at least 10 cycles of CAK cell infusions. As shown in Table I, clinical responses varied among the patients. Ninety percent showed reduction in tumor markers. The best response of three patients was complete response (patients no. 13, 16, 20 and 23), although the response did not continue. Partial response was observed in 17% of the patients. Interestingly, the patients with stable disease had longer survival than those with partial response.
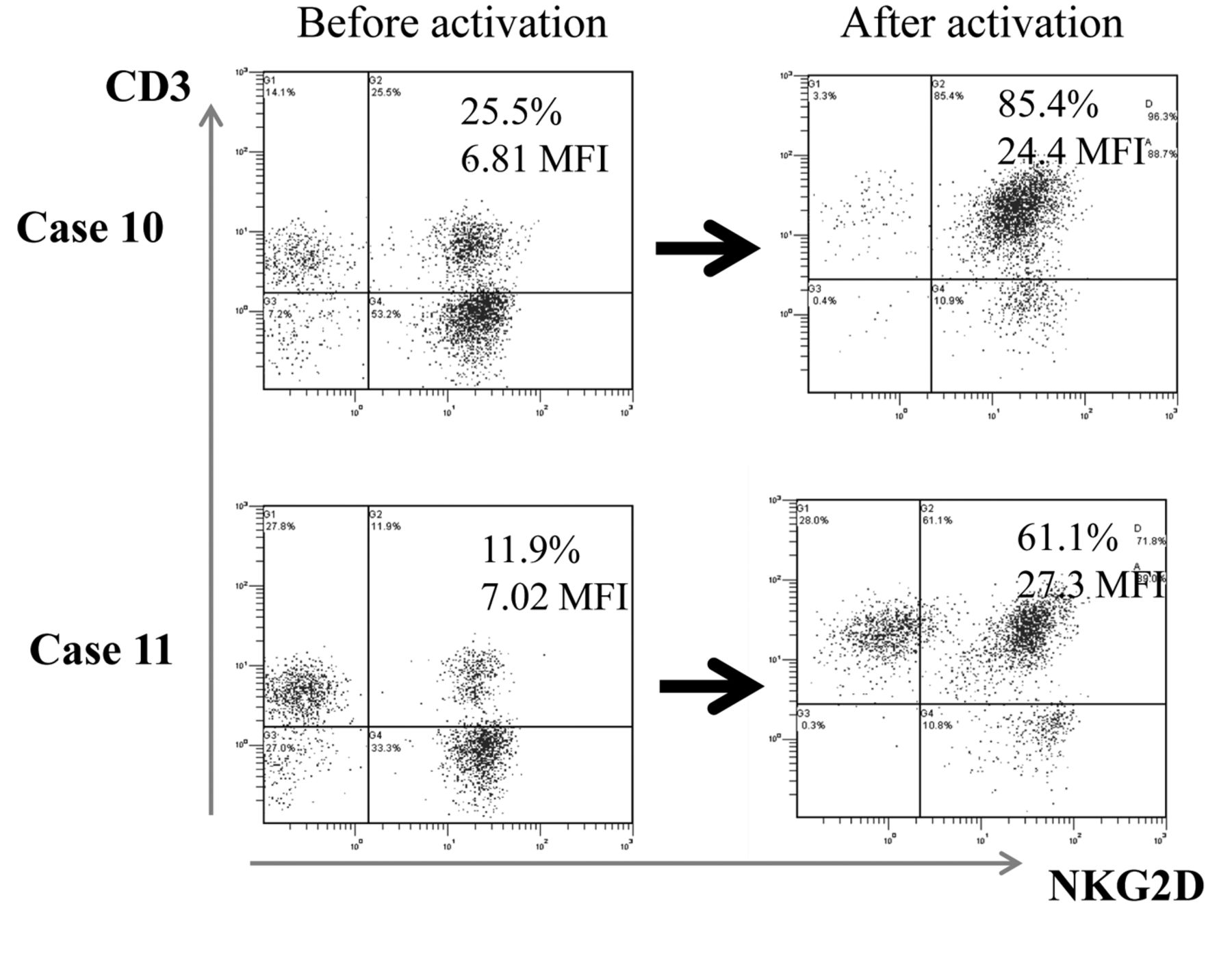
Characterization of infused CAK cells. All CAK cell batches were found to consist mainly of CD3+ NKG2Dhigh T-cells (Table II). Based on the activation of NKG2D, the NKG2Dhigh CD3+ cells constituted a median of 55% (range 36.591.6%). Both the percentage and the mean intensity of NKG2D expression significantly increased after activation: the mean percentage of NKG2D+CD3+ cells before activation was 25.1±2.34% vs. 63.9±2.97% after activation, and the mean fluorescence intensity of NKG2D before activation was 7.08±0.44 vs. 16.7±1.35 after activation (Table II and Figure 1). Two representative cytograms of NKG2D/CD3 expression are shown in Figure 2.

Expression of Natural-killer group 2, member D (NKG2D) on lymphocytes before and after activation with interleukin-2 (IL-2) and OKT3. A: Percentage of cells with positive expression of NKG2D. B: Mean fluorescence intensity of NKG2D. *p<0.01 (paired t-test).
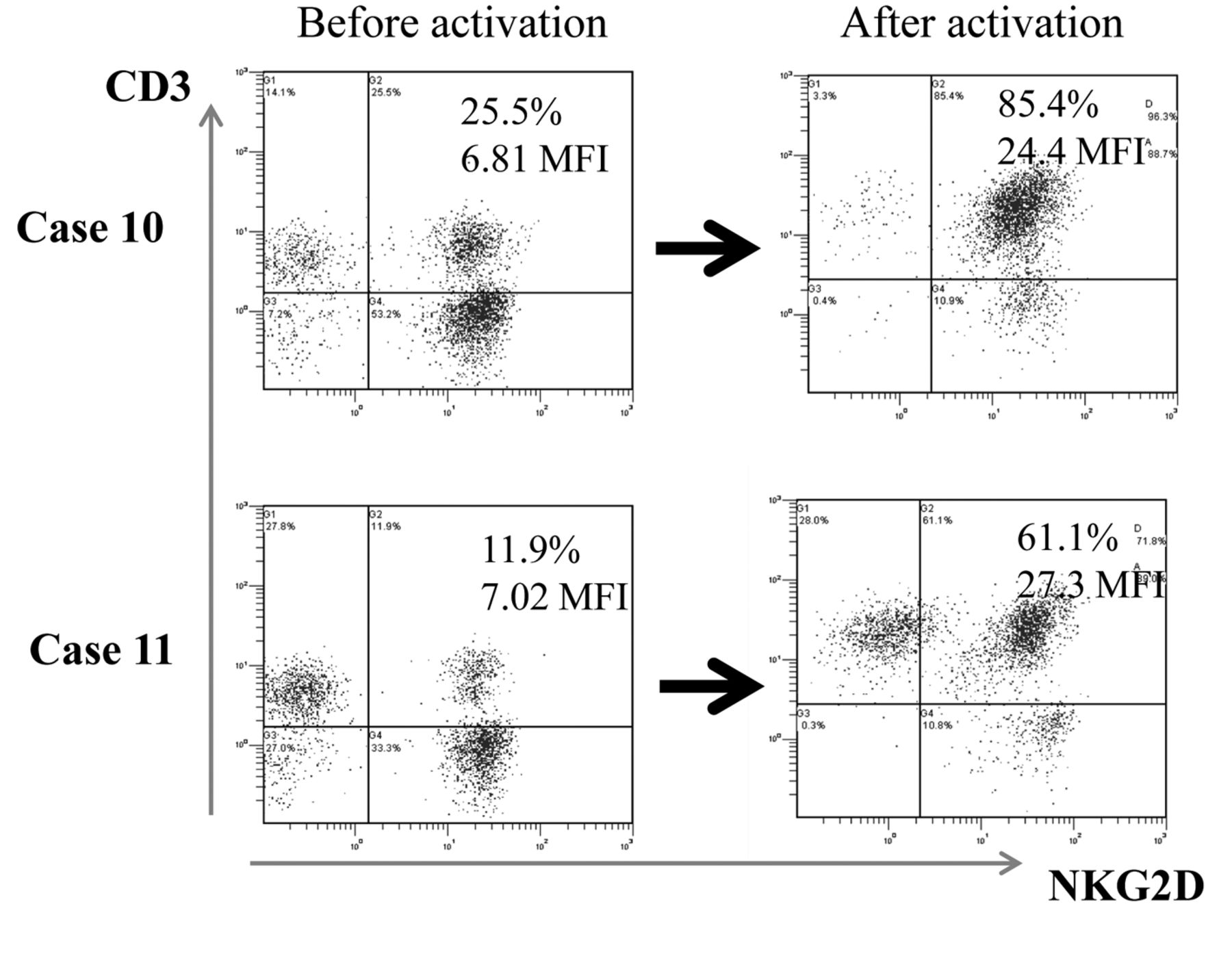
Representative histogram of Natural-killer group 2, member D (NKG2D) on lymphocytes before and after stimulation. Peripheral blood mononuclear cells derived from case 10 and case 11 were cultured in the presence of interleukin-2 and the agonistic antibody to CD3. NKG2D expression of lymphocytes before and after activation was analyzed by fluorescence activated cell sorting analysis. The notable increase in expression of NKG2D on CAK cells is shown.

Modulation of Natural-killer group 2, member D (NKG2D) ligands (major histocompatibility complex-class I-related chain (MIC)A/B, UL16-binding protein (ULBP)-1, and ULBP-3) expression on the KLM-1 cell line after gemcitabine treatment. KLM-1 cells were treated with gemcitabine at 0.1 or 1 μg/ml for 48 h, and MICA/B, ULBP1, and ULBP3 surface expression was analyzed by flow cytometry. Data are representative of three independent experiments. The inset number of each histogram indicates the mean fluorescence intensity.
Gemcitabine up-regulates NKG2D ligand expression in various carcinoma cell lines. To investigate the underlying mechanisms by which CAK cells effectively kill tumor cells after gemcitabine treatment, we first examined the expression levels of NKG2D ligands protein on the cell surface in the presence of gemcitabine using flow cytometry analysis with antibodies that recognize MICA/B, ULBP1 and ULBP3. KLM-1 cells were cultured with gemcitabine (0, 0.1 or 1 μg/ml) for 48 h prior to determination of MICA/B expression. Treatment with gemcitabine markedly augmented MICA/B protein expression on the surface of KLM-1 cells (Figure 3). Modulation of MICA/B expression on KLM-1 cells was concentration-dependent, with significant up-regulation seen in cells treated with gemcitabine concentrations of 1 μg/ml for 48 h (Figure 3). A slight increase in ULBP1 expression by KLM-1 cells was also observed after gemcitabine treatment (Figure 3). A time-course experiment of KLM-1 cells treated with 10 μg/ml gemcitabine revealed that MICA/B expression increased in a time-dependent manner. Next, we determined the effects of gemcitabine on the mRNA expression of MICB in KLM-1 cells. Quantitative real-time RT-PCR analysis revealed that MICB transcripts increased after incubation of cells with gemcitabine for 24 h (Figure 4).
Increased MICA/B expression following gemcitabine treatment enhances the susceptibility of KLM-1 cells to CAK lysis. We next evaluated the effect of gemcitabine treatment on the sensitivity of KLM-1 cells to killer cells by measuring the cytotoxicity of CAK cells against KLM-1 cells that were pre-treated with gemcitabine, using the calcein-release assay. To determine whether altered levels of MICA and MICB on tumor cells aid the recognition of KLM-1 cells by CAK cells, we measured NKG2D expression in effector CAK cells from one patient (patient 10; Figure 4). When subjected to cytotoxicity testing, we found that pretreatment with gemcitabine (0.1 or 1 μg/ml) significantly increased the susceptibility of KLM-1 cells to cytotoxicity by CAK cells (Figure 5). Furthermore, this synergistic effect of the combined gemcitabine and CAK therapy was evident in all time points (4 h, 12 h, and 24 h) studied (data not shown).

Changes in (major histocompatibility complex-class I-related chain (MIC)A/B mRNA expression in KLM-1 cells after exposure to gemcitabine. KLM-1 cells were treated with gemcitabine at the indicated doses of gemcitabine for 24 h, then analyzed for MICB mRNA expression. A: Real-time reverse transcription-Polymerase Chain Reaction plots of MICB mRNA. B: Data are expressed as the fold increase in mRNA expression in gemcitabine-treated KLM-1 cells relative to untreated cells.

Cytotoxicity of cytokine activated killer (CAK) cells with gemcitabine against KLM-1 pancreatic carcinoma cells. A: Calcein-releasing assay with imaging Lumina Vision analysis for detecting the cytotoxicity of CAK cells against KLM-1 cells following pre-treatment of the KLM-1 cells without or with gemcitabine (0, 0.1 or 1 μg/ml). The effector (CAK):target (KLM-1) cell ratios (E:T) were 20:1, 40:1 and 80:1. B: All data are expressed as the average specific cytotoxicity percentage±standard error (SE) of triplicate wells and are representative of three independent experiments. *p<0.01, Comparing gemcitabine alone and gemcitabine with CAK.
Effects of soluble MICB on CAK-mediated cytotoxicity against tumor cells. Soluble NKG2D ligands have been shown to inhibit NKG2D-mediated antitumor cell activity. There is considerable evidence showing that shedding of MIC ligands in a soluble form represents a mechanism of tumor escape from NKG2D-mediated immune surveillance, and that engagement of soluble MICB with NKG2D down-regulates NKG2D expression on CD8+ T-cells to suppress T-cell activation (6). Therefore, we investigated whether recombinant soluble MICB inhibits activated lymphocyte- and NK cell-mediated antitumor cytotoxicity. When NKG2D–MICB interactions were blocked with saturating amounts of recombinant soluble MICB, the killing of gemcitabine-treated tumor cells by CAK cells was significantly reduced in a dose-dependent manner (Figure 6). These results indicate that CAK-mediated cytotoxicity was at least partly dependent on the NKG2D–MICA/B system.
Soluble MICB induction is not affected by gemcitabine. Culture supernatants from KLM-1 cells (5×104 cells/ml) treated with or without gemcitabine for 48 h were analyzed for soluble MICB concentration. Concentrations of soluble MICB were not affected by gemcitabine treatment, indicating that gemcitabine did not induce shedding of soluble MICB from tumor cells (data not shown)
Case study. In Figure 7, a representative result from patient no. 23 is shown. Using CAK cells from this patient, we investigated the cytotoxicity of CAK cells with and without different doses of gemcitabine against T24 urothelial carcinoma cells, because this patient had bladder cancer with multiple liver and lymph node metastases. Gemcitabine treatment at doses of 0.1 and 1 μg/ml for 24 h induced an increased expression of MICA/B on T24 tumor cells (Figure 7A). The cytotoxicity of CAK cells with gemcitabine was significantly higher than that of CAK cells-alone and gemcitabine-alone (Figure 6B). In the case of patient no. 23, the tumors completely disappeared with the combination therapy of gemcitabine and CAK therapy (Figure 7C).
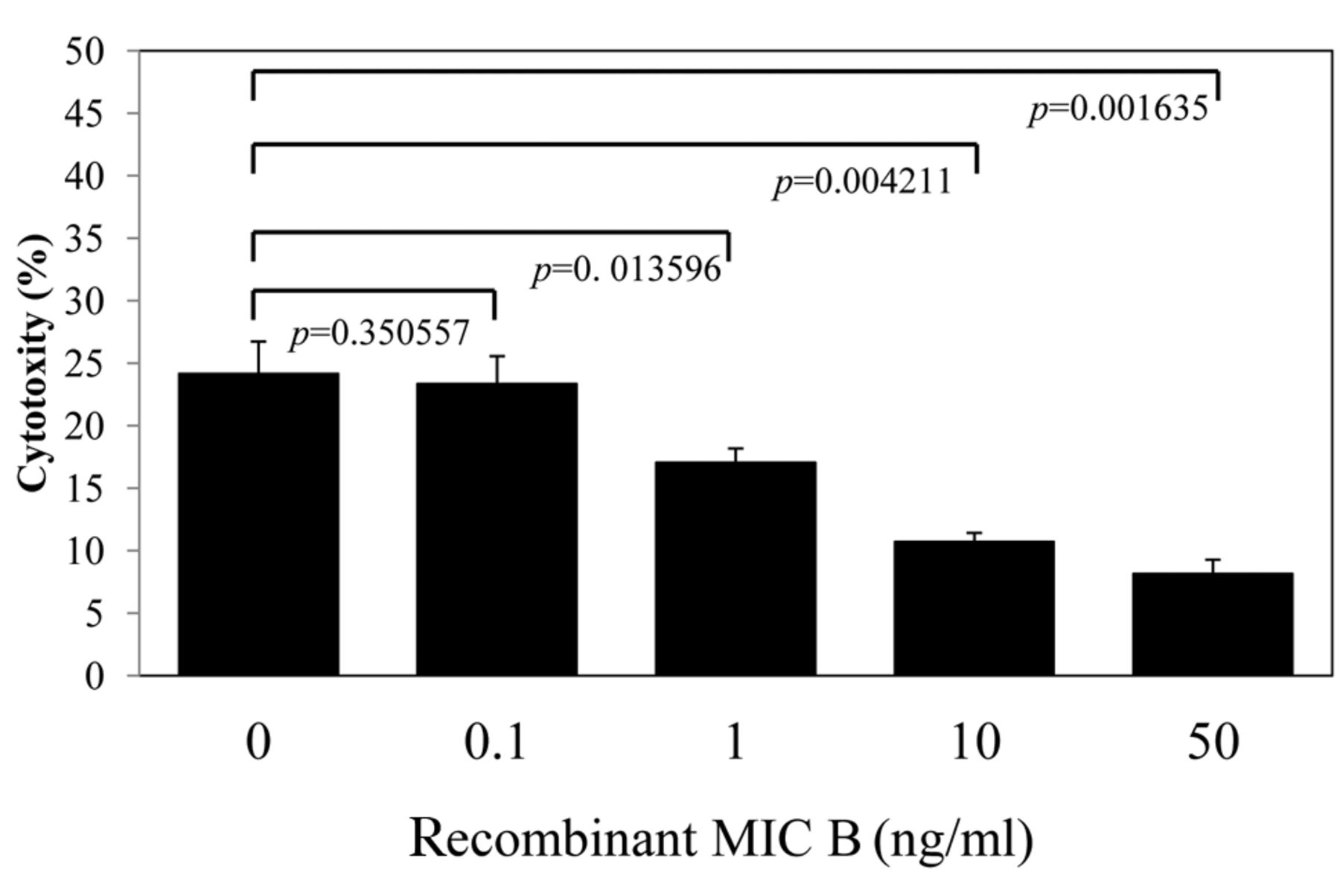
Effect of recombinant (major histocompatibility complex-class I-related chain (MIC)A/B on cytokine activated killer (CAK) cells-mediated cytotoxicity against KLM-1 cells. KLM-1 cells were incubated with or without soluble recombinant MICB for 4 h at 37°C before addition of CAK cells. Cells were incubated for another 4 h prior to performing the adherent target detachment assay as described in Patients and Methods. All data are expressed as the average specific cytotoxicity percentage±SE of triplicate wells and are representative of three independent experiments. p-Values comparing control (no soluble MICB) to treated groups are shown.
Discussion
The results of this retrospective study show that adoptive transfer of PBMC-derived CAK cells in combination with gemcitabine for more than six months is safe, as it induced disease control (complete/partial response and stable disease) in the treated patients displaying progressive metastatic solid tumors before treatment, without any major adverse events related to the infused CAK cells. To unravel potential mechanisms underlying the clinical responsiveness of patients, we performed a detailed characterization of the infused T-cell batches and observed that the cytotoxic activity of CAK cells significantly increased after gemcitabine pretreatment.
In the basic research part of this study, we showed that the surface protein expression of MICA/B and the mRNA level of MICB increased after exposure of pancreatic carcinoma cells (KLM-1) to gemcitabine. We also found that gemcitabine did not increase MICB shedding from KLM-1 cells (data not shown). Up-regulation of the NKG2D ligand, MICB, has been reported to occur following severe stress such as heat shock, bacterial or viral infection, DNA damage, oxidative stress, and treatment with retinoic acid or histone deacetylase inhibitors (8, 21). In our previous study, we have reported that gemcitabine induced an increase in MICA/B expression on HepG2 hepatocellular carcinoma cells (9). Although we did not examine this, an increase in MICA/B expression by KLM-1 cells or T24 cells may be related to ataxia-telangiectasia, mutated (ATM) and ATR (ATM and Rad 3-related) protein activation.

Experimental and clinical findings of patient no. 23. A: Effect of gemcitabine on MICA/B expression on urothelial carcinoma T24 cells. T24 tumor cells were treated with or without gemcitabine for 24 h and analyzed for MICA/B expression with FACS analysis. B: Effect of gemcitabine with cytokine-activated killer (CAK) cells on urothelial carcinoma T24 cells. T24 tumor cells were treated with CAK alone and with various doses of gemicitabine pretreatment. C: Clinical findings of patient no. 23. Computed tomography of liver metastasis and lymph node metastasis before (December 2008) and after (March 2011) therapy.
We have shown that the gemcitabine-induced effects on MICA/B expression enhanced the susceptibility of tumor cells to the cytotoxicity of CAK cells. We conclude from this result that the interaction between NKG2D and its ligands, MICA and MICB, plays a role in CAK-mediated lysis of different carcinoma cells, and that the increased susceptibility of gemcitabine-treated cancer cells to CAK cell-induced cytotoxicity may be mediated by up-regulation of NKG2D ligands. Given the extensive distribution of NKG2D in immune cells (e.g., NK, T- and γδT-cells) and the antitumor significance of the interactions between NKG2D and its cognate ligands, it is reasonable to predict that gemcitabine may augment CAK immunotherapies.
The CAK cells used in this study were a heterogeneous population including activated NK cells and T-cells. The cytotoxicity of both cell populations is at least partly dependent on the NKG2D–MICA/B systems. Importantly, NKG2D can help NK cells to overcome inhibitory signals, and substantially enhances the cytotoxicity of activated T-cells against tumor cells independently of any interaction with TcR class I (22). Activated expanded CD8+ T-cells can also lyse tumor cells through the NKG2D systems independently of the TcR class I system (23-25), demonstrating that NK, γδT-cells and activated expanded CD8+ T-cells can all act via the NKG2D system and independently of interactions with the TcR-HLA class I system. MICA expression on tumor cells has been demonstrated to be related to the efficacy of activated T-cell therapy in patients with non-small cell lung cancer (26).
In conclusion, we have shown that the combination of gemcitabine with CAK cell immunotherapy has clinical significance for therapy of patients with various disseminated carcinomas. The results of this retrospective study warrant a randomized clinical study on the efficacy of the combination of CAK therapy with gemcitabine.
Footnotes
-
This article is freely accessible online.
- Received April 4, 2014.
- Revision received June 9, 2014.
- Accepted June 10, 2014.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Mutant KRAS Promotes NKG2D+ T Cell Infiltration and CD155 Dependent Immune Evasion
- Efficacy and biomarker analysis of nivolumab plus gemcitabine and cisplatin in patients with unresectable or metastatic biliary tract cancers: results from a phase II study
- Cumulus Cells Are Potential Candidates for Cell Therapy
- T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells
- Memory T Cells Expressing an NKG2D-CAR Efficiently Target Osteosarcoma Cells
- Resveratrol Overcomes Cellular Resistance to Vemurafenib Through Dephosphorylation of AKT in BRAF-mutated Melanoma Cells