


Abstract
Background/Aim: Vaccination with fusions of dendritic cells (DCs) and mucin-1 (MUC1)-positive tumor cells (FC/MUC1) induces MUC1-specific antitumor immunity. However, little is known about the function of Cluster of Differentiation (CD)4 T-cells primed with FC/MUC1 in MUC1 transgenic (MUC1.Tg) mice. Materials and Methods: CD4 T-cells primed with FC/MUC1 were analyzed by flow cytometry. Antitumor immunity by adoptive transfer of primed CD4 T-cells in Rag2−/− mice was assessed. Results: The effector and memory T-cells generated with FC/MUC1 were crucial to maintenance of long-term antitumor immunity. MUC1-8-mer peptide SAPDTRPA presented by FC/MUC1 was recognized by CD4 and CD8 T-cells. A subset of primed CD4 T-cells possessed cytotoxicity to lyse major histocompatibility complex (MHC) class I and MUC1 positive tumor cells. Interestingly, adoptive transfer of primed CD4 T-cells prevented lung metastasis in Rag2−/− mice. Conclusion: CD4 T-cells primed by FC/MUC1 play direct role in antitumor immunity.
Cluster of Differentiation (CD)4 T-cells recognize peptides presented by major histocompatibility complex (MHC) class II molecules, whereas CD8 T-cells recognize peptides presented by MHC class I molecules. However, such restriction is not absolute as CD4 T-cells reactive to antigens presented by MHC class I molecules have been identified in patients with melanoma (1); their physiological relevance, however, remains unknown.
Mucin-1 (MUC1), a transmembrane mucin, is a dominant tumor antigen overexpressed on adenocarcinoma cells (2) and consists of variable numbers of 20-amino-acid tandem repeats (VNTR) and carbohydrates. MUC1 is expressed by most epithelial carcinomas (3). MUC1, expressed by tumors, exhibits aberrant glycosylation patterns (4). The shorter carbohydrate side chains in mucins produced by malignant cells can potentially expose normally cryptic carbohydrate, peptide or glycopeptide epitopes (5). The absence of glycosylation at certain sites in tumor mucins also leads to unmasking of specific epitopes concealed in completely glycosylated mucins (6). Cytotoxic T lymphocytes (CTLs) can be induced against the backbone of MUC1 presented by most murine H-2 and human leukocyte antigen (HLA) alleles (7). In addition, peptides and glycopeptides from MUC1 bind to both MHC class I and II (I/II) and are recognized by CD8 and CD4 T-cells (5, 8, 9). Moreover, both MHC class I/II-restricted epitopes have been found within a single peptide from MUC1 (10).
In the present study, we evaluated CD4 T-cells primed by vaccination with fusion cells generated from dendritic cells (DCs) and MUC1-positive murine tumor cells (FC/MUC1) in MUC1 transgenic (MUC1.Tg) mice.
Materials and Methods
Mice and cells. The C57BL/6 mouse strain transgenic for human MUC1 (MUC1.Tg mice, a kind gift from Dr. Sandra J. Gendler, Mayo Clinic, Scottsdale, AZ, USA) was established as described and checked for expression of MUC1 by polymerase chain reaction (PCR) (11). Rag2−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All animal work has been conducted according to relevant national and international guidelines. In accordance with the recommendations of the Weatherall report, “The use of non-human primates in research” the following statement to this effect has been included to document the details of animal welfare and steps taken to ameliorate suffering in all work involving non-human primates. This work has been reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at Boston University school of Medicine (protocol number: AN-14045).
A human MUC1 cDNA was stably transfected into murine MC38 colon adenocarcinoma cells (MC38/MUC1) (12), murine B16 melanoma cells (B16/MUC1) (13) and B16/Ia+ tumor cells (B16/Ia+/MUC1) (B16/Ia+, a kind gift from Dr. Suzanne Ostrand-Rosenberg, University of Maryland, Baltimore, MD, USA). Cells were maintained in Dulbecco's modified Eagle's minimal essential medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. DCs were obtained from bone marrow culture of wild-type C57BL/6 mice and were cultured in 20 ng/ml recombinant murine granulocyte macrophage colony-stimulating factor (GM-CSF) (Sigma, St. Louis, MO, USA) for 5 days. The purified DCs were fused to MC38/MUC1 tumor cells in the presence of 50% polyethylene glycol (Sigma) (14) for immunization.
CD4 T-cell sorting. MUC1.Tg mice were immunized twice subcutaneously on day 0 and 7 with 5×105 FC/MUC1. In addition, 5×105 irradiated MC38/MUC1 tumor cells and PBS were used as controls. On day 14, the draining lymph node cells (LNCs) were collected and passed through nylon wool to deplete B cells and antigen-presenting cells (APC). The single lymph node T-cells (T-LNCs) were dual-stained with a fluorescein isothiocyanate (FITC)-conjugated anti-CD4 (H129, 19) monoclonal antibody (mAb) and a phycoerythrin (PE)-conjugated anti-CD8 (53-6.7) mAb (BD Pharmingen, San Diego, CA, USA) for 30 min on ice. The stained T-LNC subsets were sorted into separate tubes by MoFlo (Cytomation, Fort Collins, CO, USA) using Summit v3.0 analysis software (Cytomation). The CD4 T-cells were resorted for purification.
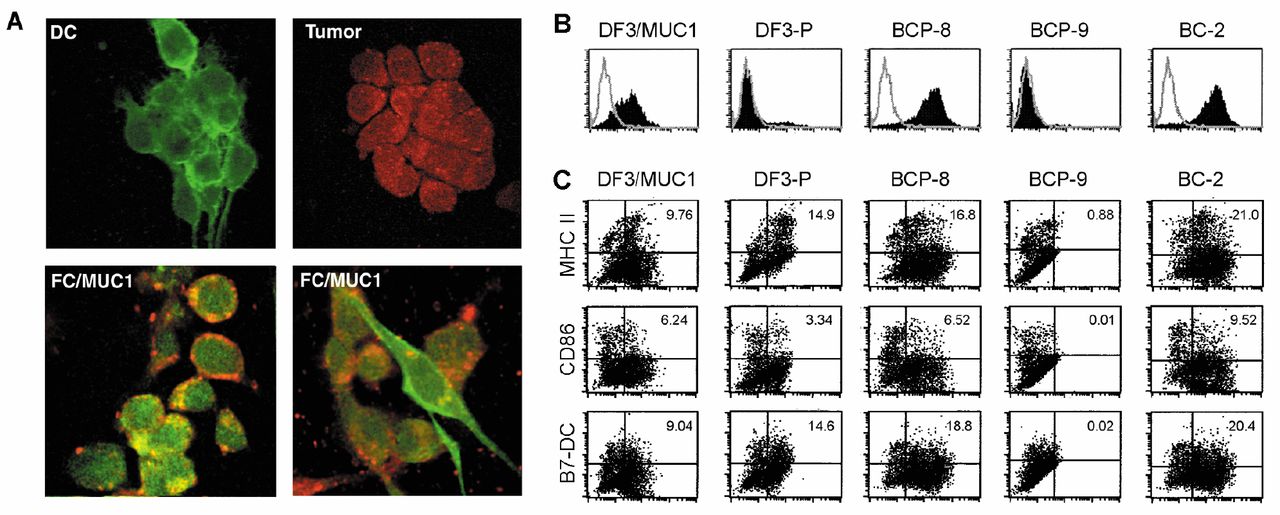
Flow cytometry. MC38/MUC1 cells were incubated with FITC-conjugated anti-DF3/MUC1 (against glycosylated MUC1) (15), DF3-P (against under-glycosylated MUC1) (16), BCP-8 (against MUC1 peptide DTR), BCP-9 (against MUC1 peptide GSTAP), BC-2 (against MUC1 peptide APDTR) mAbs (BCP-8, BCP-9 and BC-2 mAbs were supplied by Dr. Vasso Apostolopoulos) or matched isotype control IgGs. For dual expression in FC/MUC1, incubation was performed with FITC-conjugated anti-DF3/MUC1, DF3-P, BCP-8, BCP-9 or BC-2 mAbs and PE-conjugated anti-HLA-DR, CD86 or B7-DC mAbs (BD Pharmingen). T-LNCs isolated from FC/MUC1 immunized mice were passed through nylon wool, stained with FITC-conjugated anti-CD4 or CD8 mAbs and incubated with PE-conjugated MUC1-8 MHC-class-I/peptide tetramer (iTAg) (SAPDTRPA) or irrelevant tetramer (SIINFEKL) (Beckman Coulter, Chaska, MN, USA), interferon-gamma (IFN-γ), CD69, CD44, interleukin-7 receptor (IL-7R) and interleukin-15 receptor (IL-15R) (BD Pharmingen). Cells were then washed, fixed and analyzed by FACScan (BD Immunocytometry System, NJ, USA).
Confocal microscopy. DCs, MC38/MUC1 and FC/MUC1 cells were spread onto coverslips and stained with PE-labeled mAbs against MUC1 peptides DTR (BCP-8) or APDTR (BC-2) (red color) and FITC-labeled MHC class II (green color). Cells were washed, fixed and analyzed by confocal microscopy.
Cytotoxicity assay. MC38, MC38/MUC1, B16/Ia+ and B16/Ia+/MUC1 tumor cells (2×104 cells/well) were pre-labeled with 51Cr for 60 min at 37°C. Splenocytes or T-LNCs from immunized mice or sorted CD8 and re-sorted CD4 T-cells were distributed in 96-well v-bottom plates (Thermo Fisher Scientific, MA, USA) at the indicated target concentrations and co-incubated with targets for 5 h at 37°C. The supernatants were assayed for 51Cr release in a gamma counter and CTL activity was determined at various effector:target (E:T) ratios. Spontaneous release of 51Cr was assessed by incubation of targets in the absence of effectors. Maximum and total release of 51Cr were determined by incubation of targets in 0.1 % Triton X-100. The percentage of specific 51Cr release was determined by the following equation: percentage specific release=((experimental−spontaneous)/(maximum–spontaneous)) ×100.
In vivo experiments. To assess the induction of long-term antitumor immunity, MUC1.Tg mice (10 mice/group) were immunized twice with 5×105 FC/MUC1 on day 0 and 7. Control mice (6 mice/group) were immunized with 5×105 irradiated MC38/MUC1, DCs mixed with MC38/MUC1 or PBS on day 0 and 7. On day 14, mice were challenged with 5×105 MC38/MUC1. Tumor incidence and volume were determined on day 30. Mice free of tumors were re-challenged with 5×105 MC38/MUC1 on day 31. Six out of 8 (2 out of 10 surviving mice were sacrificed for CTL assays) FC/MUC1 immunized mice free of tumors were re-challenged with MC38/MUC1 on day 31 (6 mice) and the remaining 2 mice served as controls. Tumor incidence and volume were determined again on day 60 (only 5 mice remained at the end of the experiment for assessment of tumor incidence because 1 mouse was sacrificed for a CTL assay on day 38).
To assess the antitumor immunity of primed CD4 T-cells in vivo, Rag2−/− mice were inoculated with 5×104 MC38/MUC1 in the mammary fat pad. Eight days after tumor inoculation, mice were injected intravenously with 2.5×106 re-sorted CD4 T-cells from the immunized MUC1.Tg mice. The mice were monitored for tumor development. Tumors were harvested and prepared for immunohistochemistry staining as previously described (17). Lung and LN metastases were measured by a clonogenic assay as previously described (18). The number of tumor colonies in lung and draining lymph node (LN) cultures from mice with each treatment were counted.
Results
Long-term antitumor immunity induced by FCMUC1. To determine whether long-term antitumor immunity was generated by the FC/MUC1 vaccines, MUC1.Tg mice were immunized twice with FC/MUC1 and challenged with MC38/MUC1. All mice immunized with either irradiated MC38/MUC1 (6/6), DCs mixed with MC38/MUC1 (6/6) or PBS (6/6) developed tumors, whereas none (0/10) of the mice immunized with FC/MUC1 developed tumors by day 30 (Figure 1A). Moreover, mice free of tumors were re-challenged with MC38/MUC1 on day 31 and were monitored for a further 30 days; none of the mice (0/5) developed tumors (Figure 1A). These findings indicate that antitumor immunity can be maintained by long-term following an initial vaccination with FC/MUC1. Importantly, CTL activity against MUC1-positive tumor cells at 100:1 was 58.9% and 25.6% on days 15 and 30, respectively (Figure 1B). Interestingly, CTL activity increased to 48.6 % on day 38, 7 days after re-challenge with MUC1-positive tumor cells and remained between 17.8 and 28.0 % on day 60 (Figure 1B). These results suggest that the initial immunization with FC/MUC1 induces MUC1-specific CTLs that are maintained at lower levels after clearance of MUC1-positive tumors but at levels sufficient to protect the mice against re-challenge with the same tumor cells (Figure 1B). To further determine the induction of effector and memory T-cells by FC/MUC1, T-LNCs freshly isolated from MUC1.Tg mice immunized with FC/MUC1 were stained with a panel of mAbs and analyzed by FACS. Figure 1C shows that immunization with FC/MUC1 activated a subset of CD4 and CD8 T-cells, with up-regulation of IFN-γ, CD69, CD44 and IL-15R. Interestingly, T-cells expressing IL-7R, CD44 and IL-15R increased on day 60 after immunization, suggesting the presence of memory T-cells. Hence, FC/MUC1 immunization leads to the generation of effector and memory T-cells in MUC1.Tg mice. These memory T-cells can be expanded quickly after re-encounter with the antigen without the need for additional immunization. The quality of T-cells generated by the initial immunization is crucial to the magnitude and maintenance of antitumor immunity.

Effective and long-term antitumor immunity induced by FC/MUC1. (A) Tumor volume and incidence of MUC1.Tg mice immunized twice with FC/MUC1 cells (○) on days 0 and 7 are shown. Control mice were immunized twice with irradiated MC38/MUC1 (▵), DCs mixed with MC38/MUC1 (⋄) or PBS (□). (B) Splenocytes were collected on days 15 and 30 from MUC1.Tg mice immunized with FC/MUC1 (○), DCs mixed with MC38/MUC1 (⋄), irradiated MC38/MUC1 (▵) or PBS (□). Splenocytes were also collected from FC/MUC-immunized MUC1.Tg mice re-challenged with MC38/MUC1 tumor cells (●, 1 mouse on day 38 and 5 mice on day 60) or without tumor re-challenge (○, 1 mouse each on day 38 and 60). CTL activity was determined at the indicated E:T ratios. (C) T-LNCs freshly isolated from FC/MUC-immunized MUC1.Tg mice on day 15, 38 and 60 were stained with indicated mAbs. The number in the right upper quadrant is the % of double-positive T-cells.

Expression of MUC1 peptides and DC-derived molecules. (A) FC/MUC1, DCs and MC38/MUC1 cells were stained with mAbs against MUC1 peptides DTR (BCP-8) or APDTR (BC-2) (red color) and MHC class II (green color) and analyzed by confocal microscopy. (B) MC38/MUC1 cells were stained with mAbs DF3/MUC1, DF3-P, BCP-8, BCP-9 or BC-2. (C) FC/MUC1 cells were double stained with the mAbs DF3/MUC1, DF3-P, BCP-8, BCP-9, BC-2 and M5/114 (anti-MHC class II), GL1 (anti-CD86) or TY25 (anti-B7-DC).
Presentation of antigenic peptides in the context of DC-derived molecules by FC/MUC1. MC38/MUC1 stained positively for DF3/MUC1, BCP-8 and BC-2, but not for DF3-P and BCP-9, whereas DCs stained positively for MHC class II (Figure 2A and B). By contrast, FC/MUC1 cells were positive for DF3/MUC1, DF3-P, BCP-8 and BC-2, but no cells were positive for BCP-9 (Figure 2B). The co-expression of MUC1 peptides and MHC class II, CD86 and B7-DC on the surface of FC/MUC1 suggests that the fusion cells possess the ability to present MUC1 epitopes in the context of MHC and co-stimulatory molecules (Figure 2C). Interestingly, FC/MUC1 were positive for DF3-P, a mAb against underglycosylated MUC1, suggesting a deglycosylation process after fusion (Figure 2C).
Effective presentation of a dual epitope of MUC1 by fusion cells against CD4 and CD8 T-cells. To assess the MUC1-specific T-cells, the MUC1-8 iTAg tetramer was used. A subset of CD8 T-cells was positive for this MUC1-8 iTAg in FC/MUC1 immunized MUC1.Tg mice (Figure 3A). Surprisingly, a subset of CD4 T-cells were also positive for MUC1-8 iTAg, but not for the irrelevant tetramer (Figure 3A). These results suggest that MUC1-8-mer peptide SAPDTRPA is a dual epitope in FC/MUC1, presented to both CD4 and CD8 T-cells.
To further determine the cytotoxicity of the T-cells induced by the FC/MUC1 vaccines, we measured the CTL activity of sorted CD4 and CD8 cells against MHC class I- or II-positive targets. As expected, sorted CD8 T-cells lysed MHC class I and MUC1-positive targets, while the killing of MHC class II-positive targets was minimal (Figure 3B, right panel). In contrast, re-sorted CD4 T-cells with a purity of more than 99.8 % lysed both class I and II as well as MUC1-positive targets (Figure 3B, left panel). Killing of MUC1-negative targets was minimal (Figure 3B). These results indicate that T-cells induced by FC/MUC1 show robust killing of tumor cells and that CD4 T-cells can recognize and react with a MUC1-derived peptide bound to both MHC class I and II molecules.

T-cell response induced by FC/MUC1 cells. (A) T-LNCs were isolated from MUC1.Tg mice immunized twice with FC/MUC1, irradiated MC38/MUC1 or injected with PBS, stained with PE-conjugated MUC1-8 iTAg or irrelevant iTAg and FITC-conjugated anti-CD4 or CD8 mAbs. (B) Re-sorted CD4 (left panel) and sorted CD8 (right panel) T-cells were cultured with MUC1 antigen and DCs. On day 4, T-cells were incubated with 51Cr-labeled MC38 (○, MHC class-I-positive and MUC1-negative), MC38/MUC1 (●, MHC class-I-positive and MUC1-positive), B16/Ia+ (□, MHC class-II-positive and MUC1-negative) and B16/Ia+/MUC1 (▪, MHC class-II-positive and MUC1-positive) cells. CTL activity was determined at the indicated E:T ratios. Similar results were obtained in two independent experiments.
Antitumor immunity of CD4 T-cells in vivo. To assess the antitumor immunity of primed CD4 T-cells, Rag2−/− mice were injected with 5×104 MC38/MUC1 cells in the mammary fat pad. Rag2−/− mice lack natural killer T (NKT), T and B cells due to an impairment of T-cell receptor (TCR) rearrangement caused by constitutive deletion of the rag-2 gene. If left without adoptive immunotherapy, all mice developed tumors (Figure 4A). In contrast, there was no detectable tumor formation in 2 of 3 Rag2−/− mice that received CD4 T-cells from immunized MUC1.Tg mice (Figure 4B). Tumor growth delay was observed in the remaining Rag2−/− mouse that received CD4 T-cells from FC/MUC1 immunized MUC1.Tg mice. In addition, the adoptive transfer of CD4 T-cells from FC/MUC1 immunized MUC1.Tg mice eliminated lung metastasis as determined by a clonogenic assay (Figure 4C, upper panel). A few tumor colonies were observed in cultures of draining LNs from the one mouse that developed a primary tumor (Figure 4C, lower panel). It should be noted that the tumors had already metastasized to the lungs and draining LNs of the untreated Rag2−/− mice by day 8 after tumor inoculation. These results are consistent with the CTL activity at the end of the experiment. The CTL activity from Rag2−/− mice transferred with CD4 T-cells from MUC1.Tg mice against MC38/MUC1 was almost double that of control mice (Figure 4D). These results indicate that primed CD4 T-cells from FC/MUC1 vaccinated MUC1.Tg mice significantly inhibit tumor growth and eliminate metastases in Rag2−/− mice, suggesting that CD4 T-cells play a significant role in antitumor immunity.
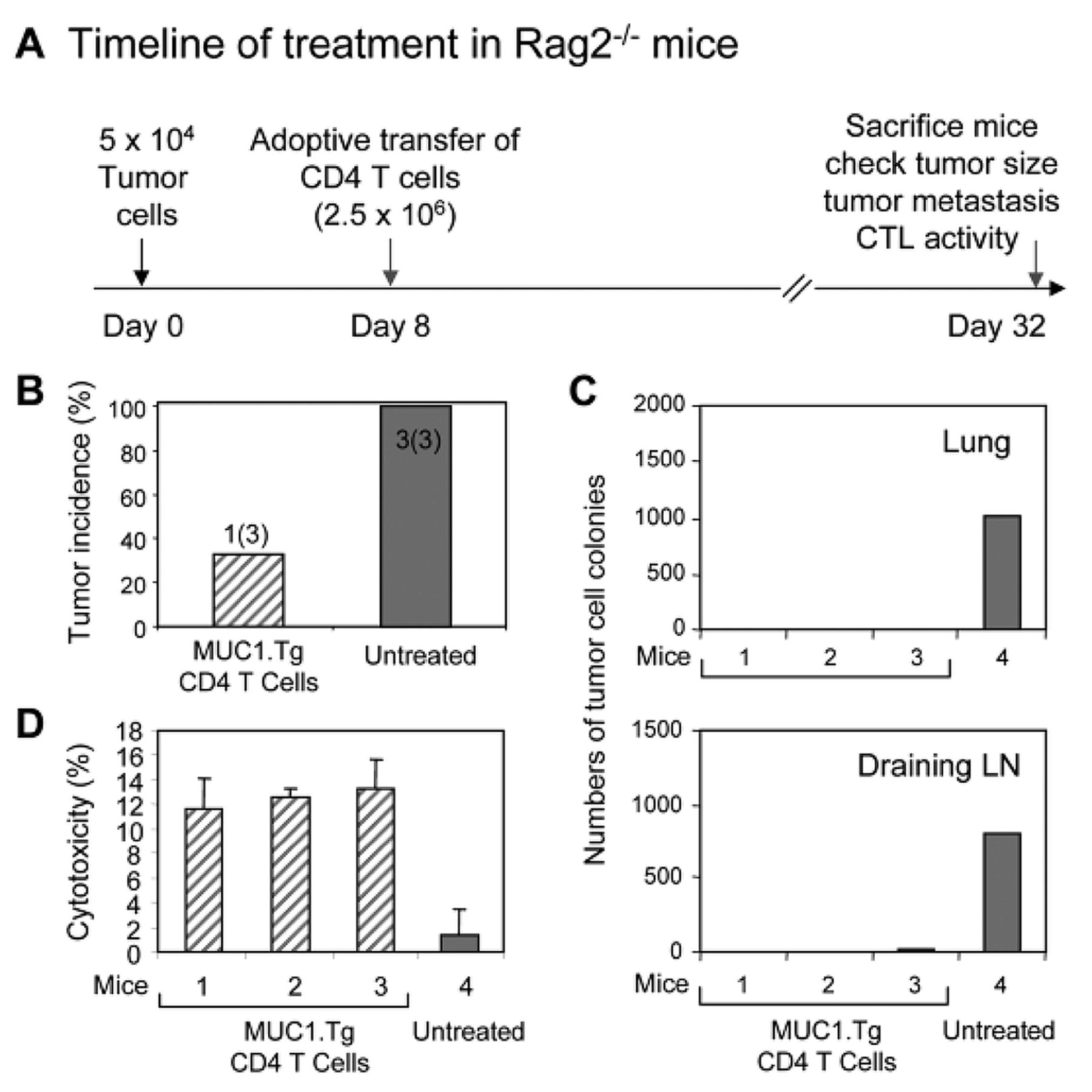
Adoptive antitumor immunity. (A) Groups of Rag2−/− mice (3 mice) were inoculated with MC38/MUC1 cells in the mammary fat pad. Eight days after tumor inoculation, the mice were treated with reselected CD4 T-cells from MUC1.Tg mice immunized with FC/MUC1 or were left untreated as a control. (B) Tumor incidence was determined by palpation on day 32 after tumor inoculation. (C) Lung (upper panel) and draining LN (lower panel) dissemination were determined by clonogenic assays. The numbers of colonies in lung and LN cultures from mice with the indicated treatments are presented in the bar graph. (D) On day 32, splenocytes were isolated and CTL activity against MC38/MUC1 (MHC class I and MUC1-positive) was determined at an E:T ratio of 50:1 with 51Cr-labeled.
Discussion
Efficient antitumor immunity requires antigen-specific CD4 and CD8 T-cells. The tumor-associated antigen MUC1 is overexpressed in various hematological and epithelial malignancies (3). We, thus, used MUC1 as a tumor antigen to investigate antigen presentation and antigen-specific CTL induction by FC/MUC1. Herein, we demonstrated that FC/MUC1 immunization leads to the generation of effector and memory T-cells, which can be expanded quickly after re-encounter with MUC1-positive tumor cells without the need for additional immunization, resulting in long-term antitumor immunity. The quality of the initial immunization may be crucial to the magnitude and maintenance of MUC1-specific CD4 and CD8 T-cells at levels sufficient to protect against MUC1-positive tumors. In this context, FC/MUC1 might be suitable for T-cell activation.
DCs and MC38/MUC1 cells were negative for DF3-P, a mAb against underglycosylated MUC1; however, DF3-P was expressed in FC/MUC1. Therefore, de-glycosylation was associated with cell fusion. Previous studies have shown that de-glycosylation of MUC1 has significant implications in the immunogenicity of MUC1-expressing cells because the absence of glycosylation at certain sites in tumor mucins leads to the unmasking of specific epitopes concealed in completely glycosylated mucins (6). Therefore, fusion cells possess the essential properties of APCs and are able to process and present antigenic peptides in the context of DC-derived molecules. Indeed, the present study also shows that FC/MUC1 cells can process and present MUC1 peptides to T-cells and induce potent antitumor immunity. In our model, the MUC1-8-mer peptide SAPDTRPA presented by FC/MUC1 was recognized by a subset of CD4 and CD8 T-cells, suggesting that SAPDTRPA is a dual epitope presented to both CD4 and CD8 T-cells. Previous reports have also demonstrated that the MUC1-11-mer peptide VTSAPDTRPAP contained both MHC class I/II-restricted epitopes (10). It is likely that such a dual epitope capability is not limited to MUC1. Indeed, a 13-mer peptide from the tumor antigen NY-ESO-1, a cancer/testis antigen that is widely expressed in a number of cancers as well as in normal testis, was recognized by both NY-ESO-1-reactive CD8 and CD4 T-cell clones (19). Increasing evidence suggests that it is necessary to engage both CD4 and CD8 T-cells to develop effective cancer vaccines. Therefore, activation of CD4 and CD8 T-cells specific for MUC1 peptides with FC/MUC1 may be an attractive approach for the development of effective cancer vaccines.
CD4 T-cells are widely known as T helper cells, required for priming, generation and maintenance of CTLs (20). However, CD4 T-cells mediate tumor rejection by a number of mechanisms, including: (i) activation of eosinophils and macrophages to produce both tumoricidal superoxide and nitric oxide (21); (ii) indirect effects of IFN-γ (22); and (iii) perforin-mediated bystander killing of neighboring MHC class II-negative tumor cells (23). Consistent with these reports, our results show that CD4 T-cells induced by FC/MUC1 are capable of lysing MHC class I positive tumor cells and regress established tumors. The underlying mechanism is currently unknown. However, the possibility of direct recognition and killing of tumor cells by CD4 T-cells cannot be excluded because a subset of CD4 T-cells induced by FC/MUC1 are positive for MUC1-8 iTAg and lyse MHC class I-positive tumor cells. Indeed, cross-reactive CD4 T-cells have been reported previously (1). Together, our findings indicate that CD4 T-cells primed by FC/MUC1 play an important and direct role in antitumor immunity in vitro and in vivo.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Footnotes
-
↵* These Authors contributed equally to this study.
-
This article is freely accessible online.
-
Conflicts of Interest
The Authors declare that they have no competing interests.
- Received June 2, 2014.
- Revision received June 18, 2014.
- Accepted June 19, 2014.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Intratumoral Accumulation and Clonal Expansion May Not Be Decisive for Rejection of Allogeneic Tumors by CD8+ T-Lymphocytes
- Liver Injury After Invariant NKT Cell Activation by Free Alpha-galactosylceramide and Alpha-galactosylceramide-loaded Dendritic Cells
- Prognostic Markers for Patient Outcome Following Vaccination with Multiple MHC Class I/II-restricted WT1 Peptide-pulsed Dendritic Cells Plus Chemotherapy for Pancreatic Cancer