


Abstract
Background: The objective of the present study was to develop an efficient delivery vehicle for siRNA LOR-1284 through incorporation of proteinase K (PrK) as a means of preventing siRNA degradation by serum nucleases. Lipid nanoparticle-PrK-siRNA (LN-PrK-siRNA) complexes were synthesized and characterized. Materials and Methods: siRNA complexed with PrK and liposomes composed of dimethyldioctadecyl ammonium bromide/cholesterol/Tween 80 (60:35:5 molar ratio) were investigated for down-regulation of R2 mRNA activity in KB human carcinoma cells. Results: Treatment with LN-PrK-siRNA (30:0.3:1 molar ratio) significantly reduced levels of R2 mRNA compared to siRNA-liposomes without PrK in serum-containing medium. LN-PrK-siRNA complexes showed increased stability in serum and reduced toxicity in KB cells relative to LN-siRNA complexes. Conclusion: LN-PrK-siRNA complexes are promising delivery vehicles for siRNA.
The therapeutic potential of gene therapy has been investigated in a variety of human diseases (1-4, 7). Small interfering RNAs (siRNAs) are duplexes of oligonucleotides, each consisting of 21-23 nucleotides (5-8). siRNA LOR-1284 targets the mRNA of the R2 subunit of human ribonucleotide reductase (RNR) (9). R2 has been reported as a key regulator of the malignant potential of tumor cells (10). For siRNA to exert its therapeutic effect when administered in vivo, it requires: i) protection from rapid degradation by serum nucleases during systemic circulation, ii) translocation across endothelial barriers, iii) facilitation of cellular uptake and endosomal escape, and iv) intracellular trafficking to the cytoplasmic site of action (11-13). Indeed, naked siRNAs are rendered inactive if they are not protected against nucleases. Liposomes averaging 100 nm in diameter are typically used in non-viral siRNA delivery as a means of improving the pharmacokinetic properties of siRNA and decreasing the rate of degradation (14). Cationic lipids can form electrostatic complexes with negatively-charged siRNA (15-17) and facilitate its delivery into cancer cells. Tween 80 is a hydrophilic surfactant added to cationic lipids that promotes the formation of stable liposomes (18-19). Proteinase K (PrK) is an enzyme of eukaryotic origin and was first isolated from the fungus Tritirachium album. Structurally, it is a serine protease containing two Ca2+ binding sites. Its ability to digest even keratin distinguishes PrK from all other proteases. PrK readily hydrolyzes native proteins, thus leading to the protective effect on oligonucleotides (20). In the present study, lipid nanoparticle-PrK–siRNA complexes (LN-PrK-siRNA) were synthesized and the protective effect of PrK on siRNA was investigated. LN-PrK-siRNA was further evaluated for R2 gene down-regulation in KB cells.
Materials and Methods
Materials. DDAB, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide (MTT), cholesterol (Chol), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Tween 80 was purchased from Acros (Fair Lawn, New Jersey, USA). PrK was purchased from Fermentas (Glen Burnie, MA, USA). RPMI-1640 media, fetal bovine serum (FBS), penicillin-streptomycin (P/S), 0.05% Trypsin-EDTA solution, and TRIzol reagent were purchased from Life Technologies (Grand Island, NY, USA). All other chemicals used were of analytical grade.
Oligoucleotides. LOR-1284 sense strand (5’-GAGAGUAGGCGAGUAUCAGdTdT-3’) and antisense strand (5’-CUGAUACUCGCCUACUCUCdTdT-3) targeting the coding region of R2 mRNA (nucleotides 1284-1304) were purchased from Alpha DNA (Montreal, Quebec, Canada).
Cell culture. KB cells (a subline of HeLa cervical cancer cell line) were cultured in RPMI 1640 supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin at 37°C in a humidified incubator with 5% CO2.
Preparation of LN and LN-PrK-siRNA complexes. LNs composed of DDAB, Chol, and Tween 80 at molar ratio of 60:35:5 were added to a stirring solution of phosphate-buffered saline (PBS, pH 7.4) to form empty LN in 10% ethanol. To form LN-PrK-siRNA complexes, PrK and siRNA were combined and added to a solution of preformed LNs at a weight ratio of 0.3:1:30, PrK: siRNA: LNs with brief vortexing. Electrostatic complexes were allowed to form by leaving the formulation at room temperature for 15 min before further studies.
Size and zeta potential measurements. The size distribution of the LNs and LN-PrK-siRNA complexes was determined by dynamic light scattering using a particle sizer (NICOMP 370, Santa Barbara, CA, USA) under the volume-weighted setting. Prior to measurement, 150 μL samples of LN or LN-PrK-siRNA were diluted with PBS to 500 μL. The zeta potential of the LN and LN-PrK-siRNA complexes were measured on a ZetaPALS instrument (Brookhaven Instrument Corp, Holtsville, NY, USA). All samples were measured in triplicate.
Gel electrophoresis analysis of LN formulations. The LN-siRNA complexes were analyzed on a 1.2% agarose gel stained with ethidium bromide (0.5 μg/mL) in 1× TBE buffer (Invitrogen, Carlsbad, CA, USA) to determine the optimal lipid-to-siRNA ratio. Varying concentrations of LN were combined with 1.0μg of siRNA in a total volume of 15 μL (including 2 μL loading dye). Electrophoresis was carried out at 100V for 30 min. siRNA mobility was visualized using a UV transilluminator (Ultra-Lum, Claremont, CA, USA) and imaged using a Kodak DC 290 digital camera (Eastman Kodak, Rochester, NY, USA).
Stability assay in plasma. To evaluate the protective effect of PrK formulations in blood plasma, LN-PrK-siRNA complexes (56 μL, siRNA 0.1 μM) were mixed with 224 μL of fresh mouse plasma and incubated at 37°C for various time periods. Samples collected at each time point were stored at −80°C prior to analysis. siRNA was extracted using a 10% sodium dodecyl sulfate solution. Then 18 μL of the LN-siRNA complex containing 0.5 μg siRNA was loaded onto an agarose gel for analysis. siRNA was detected as described above in the gel electrophoresis study.
Cytotoxicity analysis. KB cells were seeded into 96-well plates at a density of 3×103 cells/well. After 24 h incubation, cells were treated with LN-siRNA and LN-PrK-siRNA complexes (siRNA 0.1 μM) in a total volume of 100 μL media supplemented with 10% FBS for 4 h, followed by additional incubation for 48 h at 37°C in 100 μL fresh culture medium. After 48 h, 20 μL of 5 mg/mL MTT stock solution was added to each well and the plate was incubated for 4 h at 37°C. Subsequently, the medium was removed and the remaining formazan crystals were dissolved in 200 μL DMSO. Cell viability was measured at 560 nm on an automated plate reader (Bio-Rad, Hercules, CA, USA).
Transfection study and RNA isolation. KB cells were seeded in 6-well plates at a density of 3×105 cells/well in 1 mL medium for 24 h at 37°C. The culture medium was removed and replaced with a medium containing LN-PrK-siRNA at different molar ratios of siRNA:PrK, 1:0.3, 1:0.5, 1:1, and 1:2, for 4 h. Transfection was carried out under serum-free conditions or at 10% FBS. After 4 h of treatment, the medium was replaced with 1 mL fresh media. After additional 48 h of incubation, the cells were lysed with 1mL TRIzol reagent for 10 min and mRNA was extracted according to the manufacturer's protocol. Extracted RNA concentrations were quantified by measuring the UV absorbance at 260 nm using a NanoDrop spectrophotometer (ND1000, NanoDrop Technologies, Inc., Wilmington, DE, USA). All transfection experiments were performed in triplicate.
Reverse transcription-polymerase chain reaction (RT-PCR). The R2 mRNA level in KB cells was evaluated using real-time qRT-PCR. 1 μg RNA at 65°C was incubated with random hexamer primers and dNTPs for 5 min, followed by cDNA synthesis using SuperScript™ III first-strand synthesis kit (Life Technologies, Carlsbad, CA, USA) in the presence of reaction buffer, dithiothreitol, MgCl2 solution, RNase OUT, and SuperScript III. Synthesis of cDNA was conducted according to the following program: incubation at 25°C/10 min, 50°C/50 min, 85°C/15 min, and 4°C/10 min in a thermal cycler (Bio-Rad, Hercules, CA, USA). The resulting cDNA was amplified by SYBR green PCR Mastermix (Applied Biosystems, Foster City, CA, USA) and specific primers. The following primer sequences were used: R2 forward primer: 5’-GCCTGGCCTCACATTTTCTAAT-3’ and R2 reverse primer: 5’-GAACATCAGGCAAGCAAAATCA-3’ and a housekeeping gene, beta-actin forward primer: 5’-TCCCTGGAGAAGAGCTACGA-3’ and beta-actin reverse primer: 5’-AGCACTGTGTTG-GCGTACAG-3’. All primers were purchased from Alpha DNA. qRT-PCR was performed under the following settings: 95°C/10 min, 35 cycles of 95°C/15 s, 60°C/30 s, 72°C/40 s. Relative gene expression values were determined by the ΔΔCT method using the StepOne v2.0 software (Applied Biosystems, Foster City, CA, USA). In all cases, R2 mRNA was normalized to the beta-actin mRNA level for each sample and all experiments were performed in triplicate.
Temperature-dependent study for in vitro activity. LN-PrK-siRNA complexes were prepared as described previously and left standing at various temperatures (4°C, 18°C, 37°C, 55°C) for 15 min before zeta potential measurement or transfection in 10% FBS supplemented medium. R2 mRNA expression was evaluated by the method described above.
Statistical analysis. Data are presented as means and standard deviations (S.D.) analyzed by a two-tailed Student's t-test using the MiniTAB software (Minitab Inc., State College, PA, USA). The cut-off of statistical significance was set at p<0.05.
Results
Characterization of LN-PrK-siRNA complexes. In order to improve the delivery and efficacy of LOR-1284, in this study PrK was added to LN-siRNA to produce a novel formulation. The siRNA was combined with PrK and then added to pre-formed empty LN to form LN-PrK-siRNA complexes. The particle size and zeta potential values of LN were determined to be 159.3±5.0 nm and 32.8±1.37 mV, respectively. LN-PrK-siRNA complexes showed a slightly increased particle size of 189.9±4.2 nm and a zeta potential of 31.35±2.06 mV. Zeta potential values using varied ratios of PrK:siRNA, 0, 0.3, 0.5, 1, 2:1 showed slight changes in potential, at +32.8, +30.2, +29.3, +28.4, and +25mV, respectively, as the PrK concentration was increased (Figure 1). Data are represented as means±S.D. of three separate experiments.
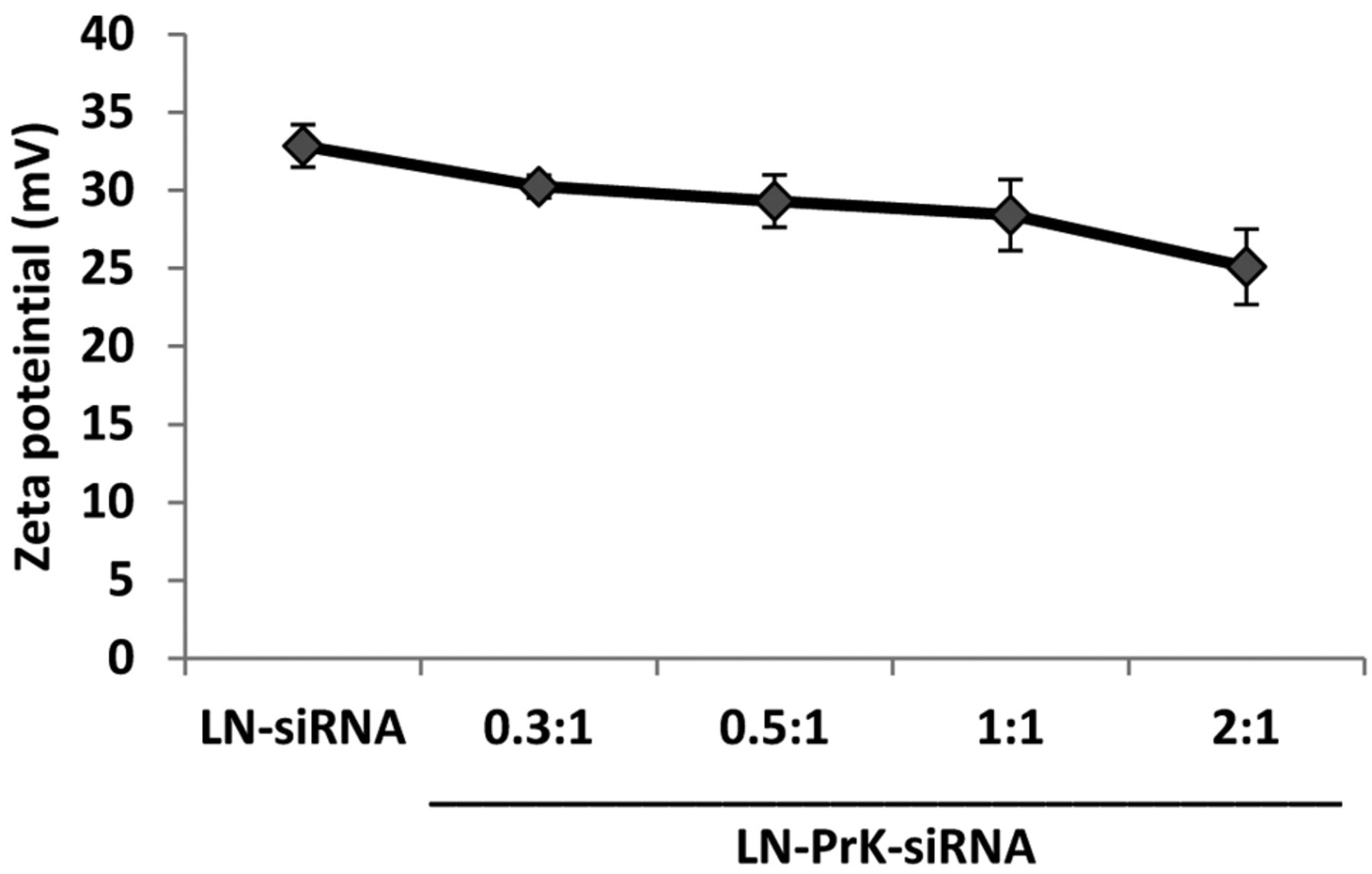
Zeta potential values of LN-siRNA and LN-PrK-siRNA with varying amounts of PrK.

Gel electrophoresis of LN-siRNA at siRNA:-liposome (w/w) ratio respectively.
Gel retardation analysis of LOR-1284 with LN. The ability of LN to form electrostatic complexes with siRNA was investigated by determining the relative mobility of the formulations during electrophoresis. LN-siRNA complexes were formed at lipid:siRNA ratios of 0, 5, 8, 10, and 20. LN-siRNA complexes were completely retarded at lipid:siRNA ratios above 10 (Figure 2).
Cytotoxicity study. The cytotoxic effect of each complex was analyzed by the MTT assay in 10% FBS-supplemented medium. As shown in Figure. 4, relative to untreated KB cells, the cell viability of LN-PrK-siRNA was 84.6±7.2% and LN-siRNA was 68.0±13.2%.
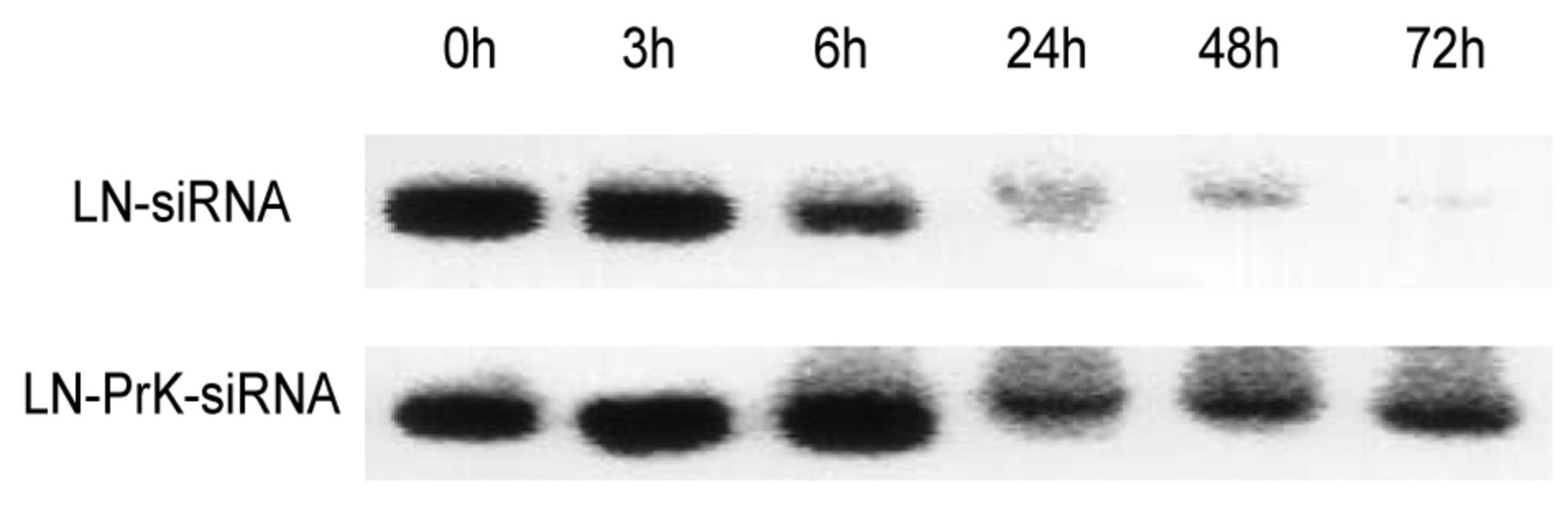
Stability study. The relative stability of LN-siRNA and LN-PrK-siRNA complexes were compared in fresh mouse plasma. The complexes were incubated at 37°C for 0, 3, 6, 24, 48 or 72 h. Figure 5 shows the protective effect of PrK on siRNA over time in the presence of serum nucleases. After treatment for 6 h, differences were observed between LN-siRNA and LN-PrK-siRNA complexes. siRNA in LN-siRNA complexes started degrading 6 h after incubation in plasma and was undetectable at 24 h. siRNA formulated with PrK, however, started degrading at 24 h, and persisted to 72 h.

Cell viability study following administration of LN-siRNA and LN-PrK-siRNA. 3.0×103 KB cells/well were seeded into a 96-well plate and incubated for 48 h at 37°C with LN-siRNA and LN-PrK-siRNA at 1:0.3 (siRNA:PrK). Untreated cells were used as a control. Cytotoxicity was assessed by MTT assay. Data represent the mean±SD (n=3). *p<0.05; comparing untreated cells and cells treated with PrK-containing complexes.

Stability study of siRNA in plasma. LN-siRNA and LN-PrK-siRNA were incubated in fresh mouse plasma at 37°C for 0, 3, 6, 24, 48, and 72 h. After incubation, siRNA was extracted by 10% SDS and detected by agarose gel electrophoresis.
R2 gene down-regulation. R2 down-regulation by siRNA was evaluated for each formulation in KB cells using real time RT-PCR analysis. Various concentrations of PrK at molar ratios 0.3:1, 0.5:1, 1:1, and 2:1 (PrK:siRNA) were investigated. To demonstrate the advantage of PrK-based LN, transfection in both serum-free and 10% FBS-containing media were studied. As shown in Figure 5, the transfection activity of LN-PrK-siRNA complexes was most profound at the PrK:siRNA molar ratio of 0.3:1 with 71±1.2% reduction in R2 gene down-regulation in medium containing 10% FBS (Figure 5b) and 54.9±0.5% down-regulation in serum-free medium (Figure 5a). In the presence of serum, enhanced R2 down-regulation mediated by LN-PrK-siRNA complexes was observed in KB cells. LN-siRNA complexes showed a down-regulation of 63.3±3.4% in serum-free medium (Figure 5a) and at 10% FBS demonstrated a reduced down-regulation to 48.4±6.1% (Figure 5b).

Effect of varying PrK concentration (0.3, 0.5, 1, 2:1, PrK:siRNA) on transfection of LOR-1284 against R2. a) without FBS, b) 10%FBS. Cont.: Untreated KB cells. R2 mRNA expression was evaluated by real-time qRT-PCR. **p<0.01,*p<0.05; of comparison of LN-siRNA and LN-PrK-siRNA at 0.3:1 (w/w, PrK:siRNA).
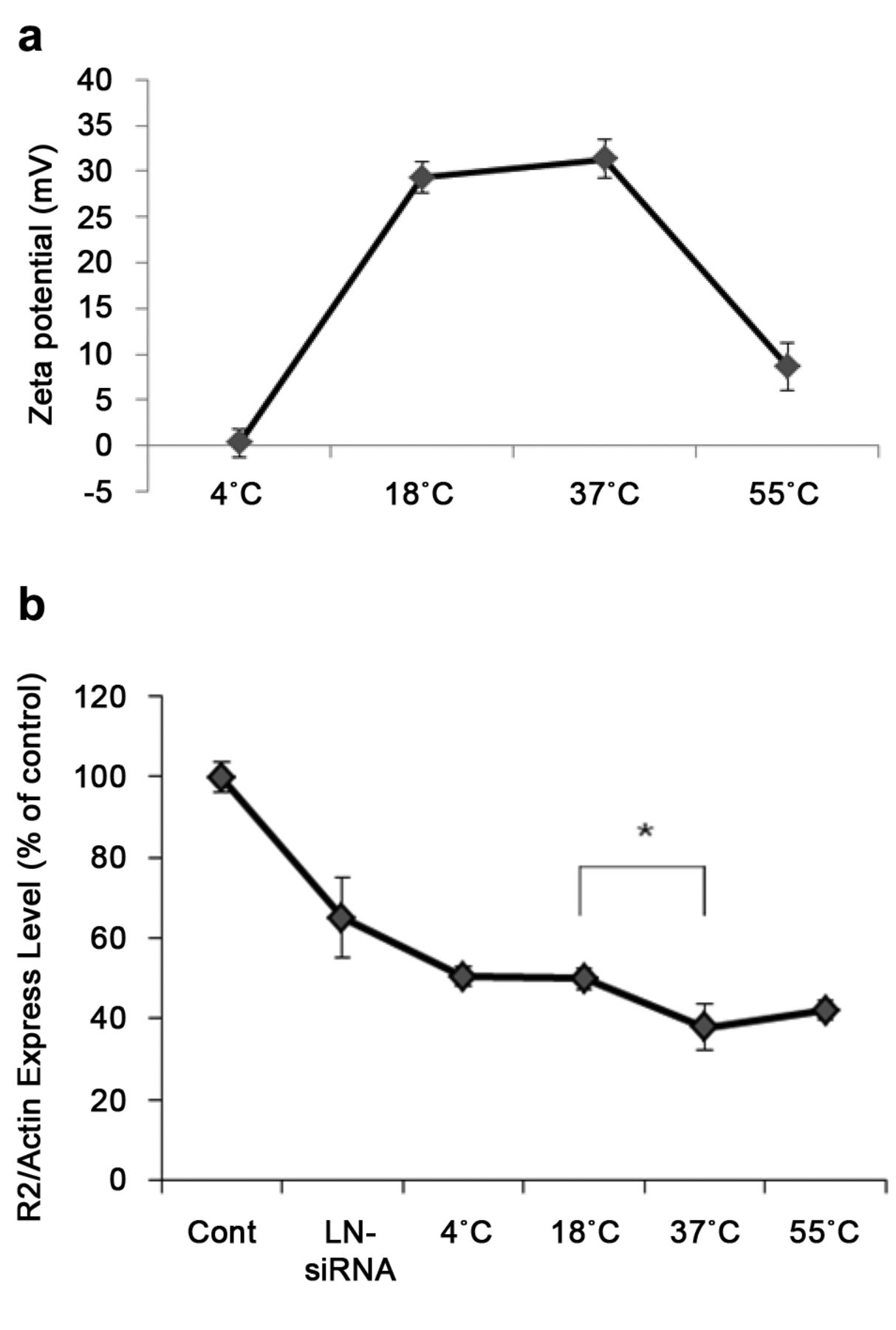
Temperature-dependent study for in vitro activity of LN-PrK-siRNA. Zeta potential values appeared to vary according to temperature: 4°C/+0.29 mV, 18°C/+29.3 mV, 37°C /+31.3 mV, and at 55°C/+8.66 mV (Figure 6a). Similar trends were observed in transfection efficiency. The effect of R2 down-regulation with varying pre-incubation temperature is shown in Figure 6b. As shown in the graph, the highest down-regulation (37%) relative to untreated cells was demonstrated by the 37°C sample. In contrast, the 4°C and 18°C samples exhibited 50.5% and 50% down-regulation respectively.

Temperature dependence study for in vitro activity of LN-PrK-siRNA. a) To make LN-PrK-siRNA complexes, PrK and siRNA were combined and mixed with preformed LNs (at a weight ratio 0.3:1:30, PrK:siRNA:LNs). Electrostatic complexes were allowed to form at various temperatures (4°C, 18°C, 37°C, 55°C) for 15 min before zeta potential measurement or treatment in KB cells. b) Effect of proteinase K on R2 down-regulation in the presence of 10% serum. LN-siRNA was left at 18°C for 15 min to form electrostatic interactions. LN-PrK-siRNA complexes were allowed to form at different temperatures for 15 min. R2 mRNA expression was evaluated by real-time qRT-PCR, *p<0.05; comparing PrK-LNs formed at 18°C and 37°C. A: Zeta potential values at various temperatures, B: R2 down-regulation at various temperatures.
Discussion and Conclusion
siRNA is a promising therapeutic modality. However, degradation by serum nucleases place critical limitations on its implementation. Lipid-based delivery systems have been utilized to improve delivery (14). Moreover, PrK provides a protective effect over RNAs by promoting hydrolytic breakdown of nucleases (20). In the present study, nanoscale LN-PrK-siRNA complexes were synthesized and effect of PrK on LN delivery efficiency was explored. The zeta potential values decreased with increasing PrK:LN siRNA: weight ratio; this may be associated with the negative charge of PrK. As shown in Figure 5, LN-siRNA complexes with PrK have a longer retention of up to 72 h in fresh plasma while LN-siRNA showed almost complete degradation within 24 h.
Figure 5 shows the transfection efficiency of LN-PrK-siRNA complexes in the absence or presence of serum. The highest R2 down-regulation was observed for the LN-PrK-siRNA complex in the presence of FBS. Among the various PrK concentrations tested, the greatest effect was found at the PrK-to-siRNA molar ratio of 0.3:1. These results demonstrate that there is an optimal ratio between siRNA1284 and PrK for delivery efficiency. At higher PrK-to-siRNA ratios there was less R2 down regulation. This is possibly due to reduced cellular uptake of the PrK-LNs because PrK is negatively charged and the uptake was dependent on the positive charge of the LNs. The enhancement in efficacy for PrK-LN at low PrK content was consistent with the results from stability study using mouse plasma where PrK exhibited a protective effect when formulated with LN-siRNA (Figure 4). The PrK-containing LNs were no better than regular LNs in serum-free media because siRNA degradation is less of a concern. The cytotoxicity of this particular PrK formulation was decreased in the presence of FBS (Figure 3). Interestingly, when the formulation was pre-incubated at 37°C, the complex demonstrated the highest zeta potential and greatest R2 down-regulation (Figure 6). Therefore, incubation temperature of the formulation may be an important factor to potentiate the therapeutic activity of PrK-based LNs. In conclusion, LN-PrK-siRNA complexes showed transfection efficiency in a PrK concentration-dependent manner. PrK confers a protective effect towards siRNA against nuclease-mediated degradation. As a result, more siRNA is delivered to the cell and is consequently able to suppress R2 expression to a greater degree. Taken together, LN-PrK-siRNA complexes are an effective and safe delivery system for siRNA therapeutics and warrant further evaluation. In addition, this novel composition should be evaluated for delivery of other oligonucleotide-based therapies such as antisense oligonucleotides, anti-miRNAs, miR mimics, and their combinations.
Footnotes
-
↵* These authors contributed equally to this work.
- Received March 26, 2014.
- Revision received May 22, 2014.
- Accepted May 23, 2014.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}