


Abstract
Aim: We evaluated the potential of TYRO3 as a therapeutic target in various types of breast cancer cell lines. Materials and Methods: The effects of TYRO3-knockdown by small interfering RNA (siRNA) on proliferation, cell-cycle distribution, and cell signaling in four estrogen receptor (ER)-positive/HER2-non-amplified (luminal-type), two ER-negative/HER2-amplified (HER2-type), and two ER-negative/HER2-non-amplified (triple negative [TN]-type) cell lines were compared. Results: Whereas TYRO3 knockdown induced the greatest proliferation suppression in luminal-type cells, and to a lesser extent in HER2-type cells, no proliferation inhibition was observed in TN-type cells. The TYRO3 siRNA-induced proliferation inhibition in luminal-type cells was observed in both estradiol (E2)-rich and -null conditions. The proliferation suppression was correlated with G0-G1/S cell-cycle arrest. Western blot analysis showed a decrease in phosphorylation of ERK1/2 or STAT3, and in cyclin D1 only in cell lines sensitive to TYRO3-knockdown. Conclusion: TYRO3 is a potential therapeutic target in breast cancer, particularly in luminal-type cells.
With 1.6 million new cases annually, breast cancer is ranked as the primary cause of cancer-related deaths among women worldwide (5). The most challenging issue in breast cancer treatment remains metastasis, because the vast majority of these cases are incurable. Treatment outcomes resulting from endocrine therapy and/or chemotherapy are not satisfactory (8, 10); therefore, development of novel treatments is needed.
For the last decade, molecularly-targeted agents, particularly those against receptor tyrosine kinases (RTKs), have been applied in the treatment of advanced solid tumors. In breast cancer, lapatinib or trastuzumab, which target gene-amplified HER2 (13, 15), have contributed to the improved treatment options for HER2-positive breast cancer as single agents or in combination with cytotoxic agents. Despite the positive outcomes induced by HER2-targeted therapies, the therapeutic potential of many other RTKs is poorly understood.
In our previous study, we showed that a novel RTK, TYRO3, regulates the proliferation of MCF-7 breast cancer cells through controlling cyclin D1 expression (3). TYRO3 belongs to the TAM (TYRO3, AXL, and MER) receptor family, which shares the same ligand, growth arrest-specific 6 (GAS6). Binding of GAS6 to these subfamily members is known to control cell proliferation and survival through the PI3K/AKT/mTOR or RAF/MEK/ERK1/2 pathways (7, 11).
Among the TAM subfamily members, AXL is the one most closely studied regarding its relevance to malignant tumors. In fact, in breast cancer, AXL has been reported to be one of the most frequently overexpressed RTKs and to have prognostic potential (6). However, few studies have reported involvement of TYRO3 in tumorigenicity, and even less its potential as a new therapeutic target. Therefore, in the present study, we explored the therapeutic roles of TYRO3 in breast cancer using a panel of eight breast cancer cell lines.
Materials and Methods
Cell culture. Four estrogen receptor (ER)-positive/HER2-non-amplified (luminal-type; CAMA-1, T47D, MCF-7, and HCC-1428), two ER-negative/HER2-amplified (HER2-type; HCC-1419 and HCC-1954), and two ER-negative/HER2-non-amplified (triple-negative [TN]-type; MDA-MB-231 and HCC-1143) breast cancer cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were maintained in RPMI 1640 (Cellgro; Mediatech, Inc., Herdon, CA, USA) supplemented with 10% FBS (Gemini-Bio-Products, Inc., Woodland, CA, USA), 100 U/mL penicillin, 100 units/mL streptomycin, and 2 mM glutamine. Estradiol (E2) was not supplemented in the maintenance media. All cells were cultured at 37°C in a humidified atmosphere with 5 % CO2 and were in logarithmic growth phase upon initiation of experiments.
TYRO3 small interfering RNA (siRNA) transfection. TYRO3 knockdown was performed using TYRO3 siRNA purchased from Invitrogen (Carlsbad, CA, USA). Optimal conditions for the gene knockdown were determined by multiple pilot experiments before transfection in accordance with the manufacturer's instructions, as previously reported (3). The sequences AACAAGUUUGGCCACGUGUGGAUGG (TYRO3 siRNA01) and AUCACUUCUUCCCACUCCAAGAUGA (TYRO3 siRNA02) were selected for efficient knockdown. To minimize the risk of off-target effects in our siRNA analysis, the stealth RNAi negative-control low GC duplex (Invitrogen) was used in each knockdown experiment.
Cell proliferation assay. MTS assay kits were purchased from Promega (Madison, WI, USA). This kit determines the number of viable cells by a colorimetric method based on the bioreduction of 3-(4,5-dimethylthiazol-2yl)-5-(3-carboxyme-thowyphenyl)-2-(4-sulfophenyl)-2H- tetrazolium (MTS) to a soluble formazan product detected spectrophotometrically at a wavelength of 490 nm. After 24 h of TYRO3 siRNA incubation, cells diluted in 10% FBS containing maintenance media without any antibiotics (160 μL/well) were plated in 96-well flat-bottom plates (day 0) (Corning, Inc., Corning, NY, USA) and a series of MTS analyses were performed from day 0 to 4 at 37°C in 5 % CO2. The number of cells required to obtain an OD of 1.3-2.2, the linear range of the assay, was determined in a preparatory experiment (data not shown); 2,000 cells/well for CAMA-1, T47D, MCF-7, HCC-1428, HCC-1419, and HCC-1954, and 1,500 cells/well for MDA-MB-231 and HCC-1143. Each experiment was repeated at least three times. Analyses of the effect of 17β-E2 (Sigma-Aldrich, St. Louis, MO, USA) were conducted using phenol-free RPMI medium (Cellgro) without antibiotics, supplemented with 10 % charcoal-stripped FBS (Invitrogen). After 24 h of TYRO3 siRNA transfection, cells were seeded in 96-well flat-bottom plates, and 10 nM of E2 with and without 1 μM of fulvestrant (Sigma-Aldrich) were added and incubated for 72 h at 37°C in 5% CO2 prior to MTS experiments.
Cell-cycle analysis. Cell-cycle analyses were performed as previously described (9). Briefly, 72 h after siRNA transfection, cells were trypsinized and cell-cycle distribution was assessed using a CycleTEST PLUS DNA Reagent Kit (Becton Dickinson [BD] Biosciences, Franklin Lakes, NJ, USA) in accordance with the manufacturer's instructions. Cells were analyzed by FACS Calibur flow cytometry (BD Biosciences) using CellQuest Pro (BD Biosciences) software and ModFit LTTM cell-cycle test software (Verity Software House Inc., Topsham, ME, USA). All experiments were repeated in triplicate.
Protein extraction and western blotting. Cells were washed once with ice-cold phosphate buffered saline and scraped immediately after adding lysis buffer (20 mM tris [pH 7.5], 150 mM sodium chloride, 2 mM EDTA, 10 % glycerol, 1 % NP40) containing protease and phosphatase inhibitors (100 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, 2 μg/mL aprotinin, 5 μg/mL leupeptin). Cell lysates were centrifuged at 14,000 × g for 10 min at 4°C to pellet-insoluble material, and the supernatant was collected as protein extract. Protein extract samples were separated using electrophoresis on 7.6 % SDS gels and transferred to polyvinylidene fluoride membranes, which were subsequently probed with primary antibodies. Phospho-AKT (Ser473), AKT, phospho-STAT3 (Tyr705) (D3A7), STAT3, TYRO3, and cyclin D1 (DCS6) were purchased from Cell Signaling Technology (Beverly, MA, USA). Phospho-ERK1/2 (pTpY185/187) and ERK1/2 (Invitrogen), anti-phosphoserine/threonine/tyrosine (Abcam, Cambridge, MA, USA), and β-actin (Sigma-Aldrich) were also purchased. Each targeted protein was detected using Amersham ECL plus Western Blotting Detection Reagents (GE Healthcare, Buckinghamshire, UK). The results are representative of at least two repeated experiments.
Immunoprecipitation. Protein A Dynabeads were purchased from Invitrogen. Following the manufacturer's instructions, 1,500 μg of protein extracts prepared as described previously were mixed with a TYRO3 antibody for 24 h at 4°C with rotation. The beads were washed three times in ice-cold washing buffer and mixed with the sample for 1 h at 4°C with rotation. After three washing cycles, the proteins were diluted in ice-cold elution buffer and LDS sample buffer and heated. The immunoprecipitation product was separated with SDS gels and subjected to western blotting, as described above.
Results
Effect of TYRO3 to proliferation of breast cancer cell lines. To determine the involvement of TYRO3 in the cell proliferation mechanism of breast cancer, we analyzed the effect of TYRO3 gene silencing on eight breast cancer cell lines (four luminal-type, two HER2-type, and two TN-type). Treatment with TYRO3 siRNA resulted in the most prominent proliferation inhibition in the tested luminal-type cell lines (Figure 1A), particularly in MCF-7, T47D, and CAMA-1. When we treated the HER2-type cell lines, a significant but less extensive proliferation inhibition than in luminal-type cells was observed (Figure 1B). In contrast, TYRO3 siRNA knockdown induced virtually no proliferation inhibition in TN-type cell lines (Figure 1C).
To further test if the TYRO3 siRNA-induced proliferation inhibition was TYRO3-specific, T47D cells were transfected with different concentrations of two TYRO3 siRNA duplexes. After 72 h, gradual inhibition of TYRO3 protein expression was obtained in a siRNA concentration-dependent manner (Figure 2). The degree of proliferation inhibition (Figure 2) was in parallel with that of TYRO3-knockdown. This finding supports that TYRO3 siRNA-induced proliferation inhibition was mediated by TYRO3 protein down-regulation.
Effect of TYRO3 knockdown on cell-cycle distribution. Since the results from the proliferation assays showed a clear impact of TYRO3 siRNA transfection by day 3 (Figure 1A and B), we decided to use 72 h as a time point to analyze the cell-cycle distribution of the siRNA-transfected cells. After exposure to TYRO3 siRNA for 72 h, an increase in the G1-G0 fraction along with a decrease in the G2-M and S fractions, indicating cell-cycle arrest at the G0-G1/S boundary, was observed only in luminal- and HER2-type cell lines (Figure 3A and B). In contrast, treatments with TYRO3 siRNA clone 1 and 2 led to little change or rather decreased the G0-G1 fraction, respectively, in TN-type cells (Figure 3B). These findings indicated that proliferation inhibition by TYRO3-knockdown was attributable to G0-G1/S cell-cycle arrest.

Impact of TYRO3 gene silencing on cell proliferation. Luminal-type (A), HER2-type (B), and TN-type (C) breast cancer cells were transfected by TYRO3 siRNA clones and negative control siRNA for 24 h. Cells were seeded in 96-well plates and serial growth assays were conducted with MTS assays from day 0 to 4. Daily optical density values shown relative to that of day 0 are shown on the y-axis. Each data point represents the mean value and standard deviation of 6-12 replicate wells.
Effect of TYRO3 knockdown on cell signaling. To explore the underlying mechanism of differential cellular responses induced by TYRO3 knockdown, we examined the downstream signaling events of TYRO3 by western blotting. In all cell lines tested, successful TYRO3 knockdown was obtained by two different siRNA clones (Figure 4A-C). In all TYRO3 siRNA-sensitive luminal- and HER2-type cell lines except HCC-1954, TYRO3 siRNAs induced down-regulation of ERK1/2 phosphorylation (Figure 4A and B). In HCC-1954, reduction in the phosphorylation of STAT3 was exclusively accompanied with that in TYRO3 (Figure 4B). In two luminal-type cell lines, decreases in cyclin D1 were also observed following treatment with TYRO3 siRNAs (Figure 4A). In contrast, no consistent change in the phosphorylation of downstream signaling molecules or cyclin D1 expression was observed in either TN-type cell line treated with TYRO3 siRNAs (Figure 4C).
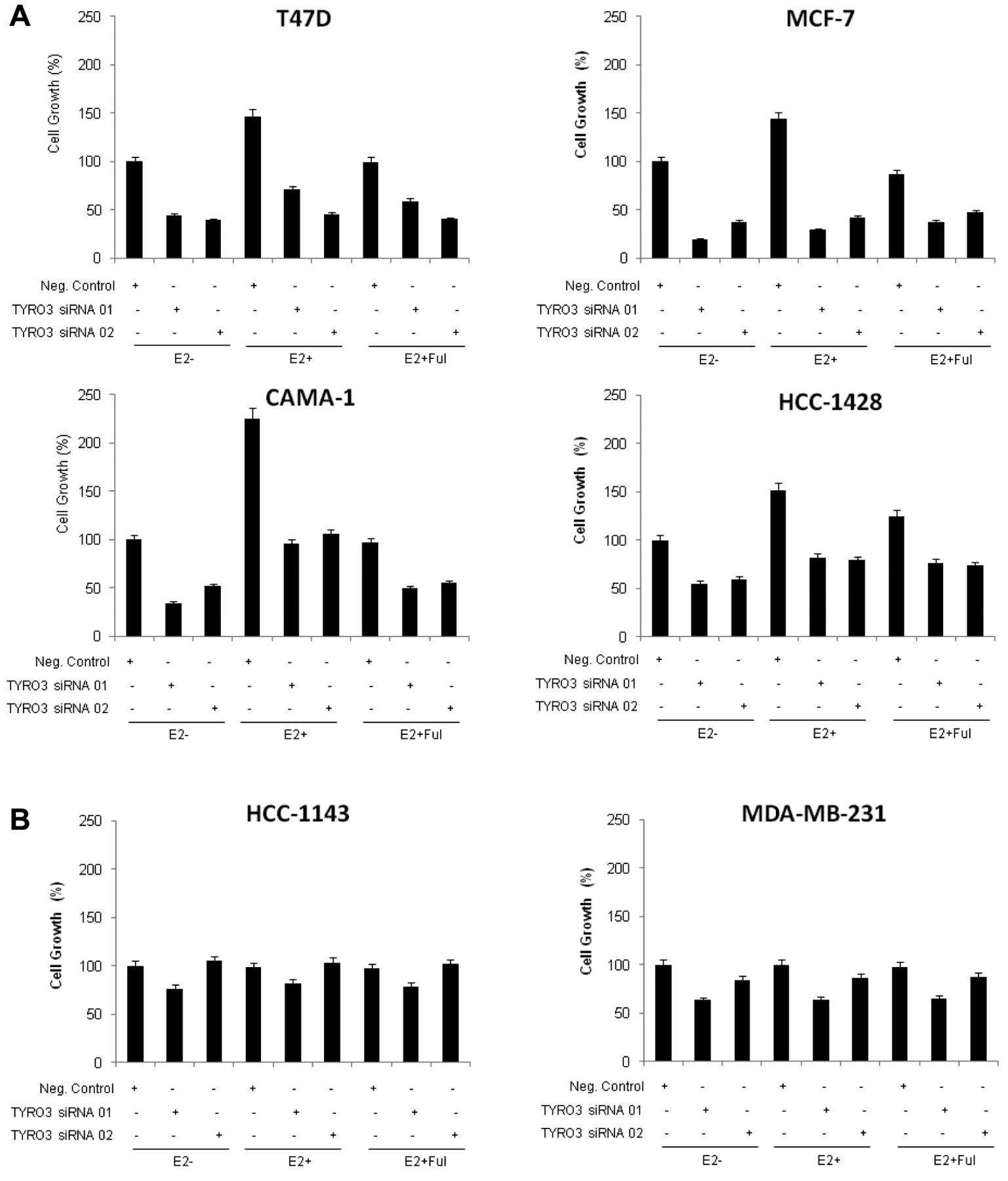
E2 Does not decrease TYRO3 siRNA sensitivity. Because we found the most promising proliferation inhibition after TYRO3 knockdown in luminal-type (ER-positive) cell lines, we decided to evaluate if cellular responses were affected by the level of co-existing E2. Cells were transfected with TYRO3 siRNA in the absence and presence of 10 nM of E2 with or without 1 μM fulvestrant, a selective ER degrader. Results from a proliferation assay showed that in all luminal-type (ER-positive) cell lines, proliferation was promoted in the E2-rich conditions compared to the E2-null conditions when treated with negative control siRNA (Figure 5A). However, the effect of E2 was negated with fulvestrant co-treatment (Figure 5A). In these luminal-type cell lines, TYRO3 siRNA inhibited cell proliferation regardless of the presence or absence of E2 or fulvestrant, suggesting that the effect of TYRO3 siRNA inhibition was independent from the estrogen system (Figure 5A). Conversely, as expected, the same treatments resulted in little change in cell proliferation in TN-type lines (Figure 5B).
Discussion
In the present study, we revealed the potential therapeutic contribution of TYRO3 inhibition in breast cancer cell lines. TYRO3 knockdown by siRNA induced the most prominent proliferation suppression in luminal-type cells, and to a lesser extent in HER2-type cells, but not in TN-type cells. This inhibitory effect was associated with G0-G1/S cell-cycle arrest and inhibition of intracellular signaling.

Correlation between TYRO3 knockdown and proliferation inhibition after siRNA transfection. T47D (luminal-type) cells were treated with different concentrations of each TYRO3 siRNA clone or negative-control siRNA for 24 h. Cells were then plated in 6-cm plates for western blotting and in 96-well plates for proliferation assays. After 72 h of siRNA transfection, TYRO3 protein expression levels were analyzed by western blotting and immunoblots were quantified by densitometry (upper panel); y-axis, TYRO3/β-actin as the average of the ratio to negative control. At the same time point, cells were subjected to MTS assays (lower panel). Optical density values are shown relative to that of the negative control siRNA and each data point represents the mean value and standard deviation of six replicate wells.
The role of TYRO3 as a therapeutic target has not previously been evaluated in detail. A recent study showed that nearly all melanoma cell lines tested overexpressed TYRO3, and knockdown of TYRO3 by short hairpin (sh)RNA led to significant cell proliferation inhibition (2), similar to our study. However, while in our study of breast cancer, proliferation inhibition appeared to be due to G0-G1/S cell-cycle arrest, in the melanoma study TYRO3 shRNA alone induced apoptosis (2). It is unknown at this point what determines cellular fate after TYRO3 knockdown.

Changes in cell-cycle distribution following treatment with TYRO3 siRNA. After 72 h of TYRO3 or negative-control siRNA transfection, cell-cycle distribution was assayed. Columns and bars represent absolute changes compared to negative control in each cell-cycle phase and standard deviations, respectively.
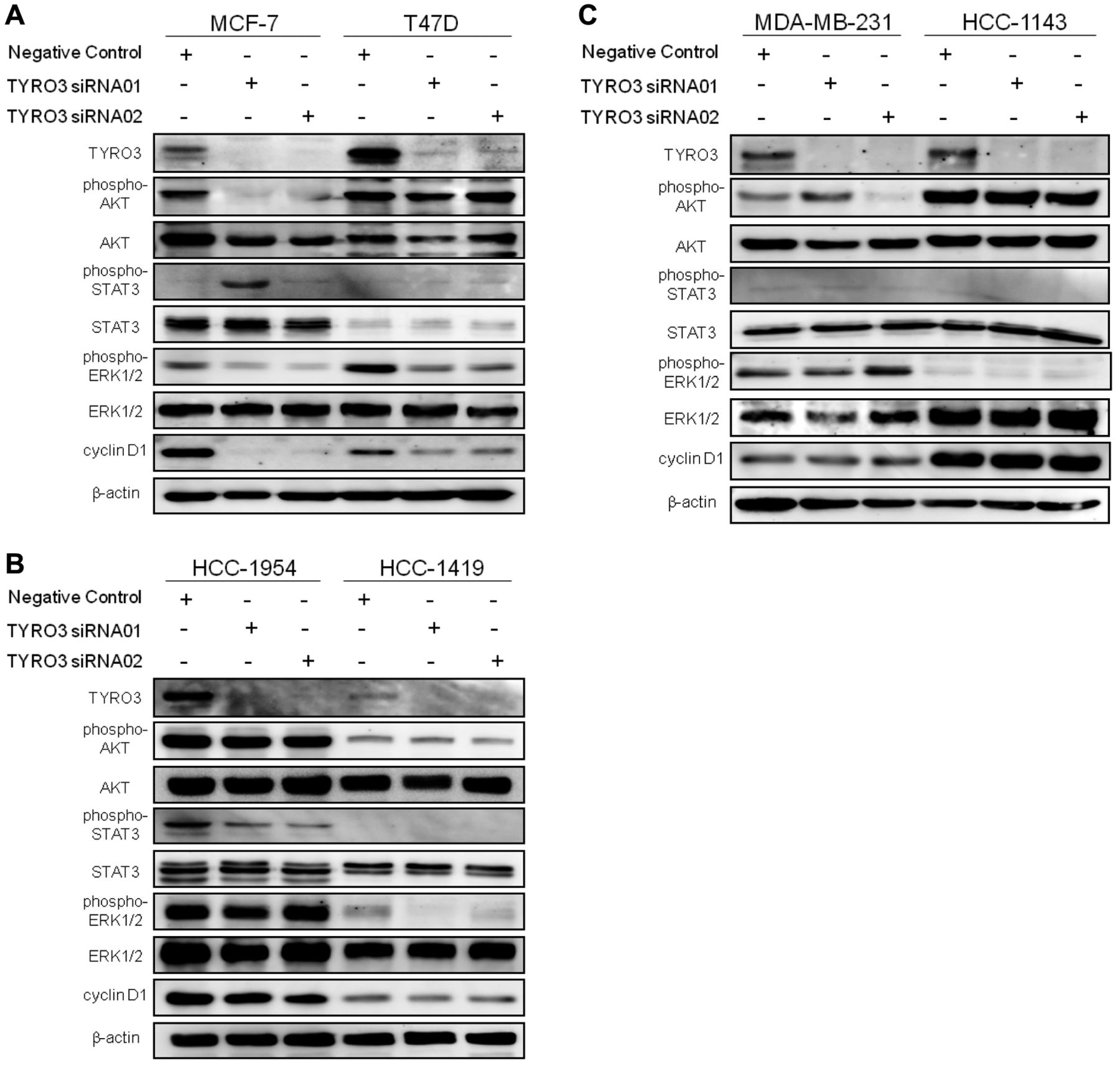
Effects of TYRO3 knockdown on phosphorylation of downstream signaling molecules. Cellular protein extracts were obtained after 72 h of siRNA transfection. The extracts were subjected to separation on SDS gels followed by immunoblotting for each protein. Blots were stripped and re-probed for β-actin, used as a loading control.
On developing new targeted-therapies, co-development of pharmacodynamic and response-predictive markers is critical. In our current study, proliferation inhibition by TYRO3 knockdown was associated with reduced phosphorylation of ERK1/2 in three out of four cell lines sensitive to TYRO3 siRNA (Figure 4A and B). Therefore, down-regulation of the phospho-ERK1/2 could be a pharmacodynamic marker. As for response-predictive markers, we investigated several candidates. The first was an intrinsic subtype of breast cancer that is widely used in the clinic; all luminal-type cell lines tested were sensitive to TYRO3 siRNA (Figure 1). The second was the TYRO3 expression level, but it could not be correlated with sensitivity to TYRO3 knockdown (Figure 4). This finding matched with the above-mentioned study of TYRO3 in melanoma, proving non-correlation between the level of TYRO3 expression and that of sensitivity to TYRO3 shRNA (2). Conversely, phosphorylation of TYRO3 was detectable in only three out of four luminal-type cell lines that were most sensitive to TYRO3 siRNA (Figure 1A and 6), which indicates its potential as a response-predictive marker, albeit unlikely universal. The third was the TYRO3 ligand GAS6; this ligand has been reported to be involved in oncogenic properties such as proliferation (12) and anti-apoptosis (12, 16) specifically in thyroid cancer (1), melanoma (2), and leiomyosarcomas (4). On screening GAS6 in the cell culture media of eight breast cancer cell lines, however, no correlation was observed between the concentration of GAS6 and TYRO3 siRNA sensitivity (data not shown).

Effects of E2 on sensitivity to TYRO3 knockdown. Luminal-type (A), and TN-type (B) breast cancer cells were transfected with TYRO3 siRNA clones and negative control siRNA for 24 h and seeded in 96-well plates. Then, 1 μM of fulvestrant and/or 10 nM 17β-E2 were applied for 72 h, and cells were subjected to MTS assays. Optical density values are shown relative to that of the negative control siRNA without E2 and each data point represents the mean value (column) and standard deviation (bar) of six replicate wells.

Phosphorylation of TYRO3 may predict sensitivity to TYRO3 inhibition. Protein extracts (1,500 μg) were immunoprecipitated with an anti-TYRO3 antibody and subjected to immunoblot assays with an anti-phosphoserine/threonine/tyrosine antibody. Phosphorylation of TYRO3 was detectable only in three luminal-type cell lines that were sensitive to TYRO3 knockdown.
Upon observing high sensitivity to TYRO3 knockdown in luminal-type cell lines, we predicted cross-talk between TYRO3 and estrogen systems. However, co-existing E2 levels did not interfere with the sensitivity of TYRO3 siRNA (Figure 5A). Although more extensive signaling analysis is required to fully-understand this issue, determinants for TYRO3 dependency in luminal-type cells may be E2 independent. Considering clinical relevance, TYRO3 inhibition might be effective in luminal-type tumors regardless of the patient's menstruation status.
There exist several limitations to our study. First, we did not investigate other possible tumorigenic properties of TYRO3, such as cellular migration and invasion, or anti-apoptosis (17). Second, we did not test the in vivo effects of TYRO3 inhibition. This was partially because no specific TYRO3-targeted drug was available. We have tested several tyrosine kinase inhibitors, including BMS777607 and vandetanib, which had been reported to have inhibitory effects on TYRO3 in cell-free conditions (14), but none of them inhibited the in vitro cell proliferation of breast cancer cell lines (data not shown). This failure might be simply due to their lack of activity against TYRO3 expression in the cell, or to complex feedback signals following off-target effects. Therefore, development of specific inhibitors against TYRO3 is necessary. In the above-mentioned melanoma study, the researchers developed three types of monoclonal antibodies against TYRO3, and the antibodies were shown to inhibit GAS6-dependent AKT activation (2).
In conclusion, TYRO3 is a potential therapeutic target in breast cancer, particularly in luminal-type cells. Future development of TYRO3-specific agents and response-predictive markers is required.
Acknowledgements
This study was supported by the Global Centers of Excellence Program (H.M.), Grant-in-Aid for Scientific Research (C) (T.M.), and a Research Grant from the Takeda Science Foundation (T.M).
Footnotes
-
Declarations of Interest
The Authors declare that they have no conflicts of interest.
- Received February 28, 2014.
- Revision received May 6, 2014.
- Accepted May 7, 2014.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}