


Abstract
Background/Aim: The current study aimed to identify an attractive target against human oral squamous cell carcinoma (OSCC). Materials and Methods: The effect of 3,3’,4’,5’-tetramethoxychalcone (TMC) on OSCC cell proliferation, cell-cycle phase distribution, expression of markers of cell apoptosis, and cell migration were analyzed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, flow cytometry, western blot, and transwell migration assay, respectively. Results: Experimental results revealed that TMC inhibited the OSCC cell proliferation (fifty percent inhibitory concentrations range=1.0-4.5 μM) by inducing G2/M phase arrest of the cell cycle. TMC caused DNA double-strand breaks, and enhanced expression of caspase-3 and -9, poly (ADP-ribose) polymerase, cytochrome c, calpain-1 and -2, phosphorylation of histone H2AX, phosphorylation of checkpoint kinases 2, p53, BCL2-antagonist/killer and BCL2-associated × protein, while reducing the mitochondrial membrane potential, and expression of B-cell lymphoma-2. In addition, TMC reduced the migration potential of OSCC cells by attenuating the C-C chemokine ligand 5/C-C chemokine receptor type 5 axis. Conclusion: These data indicate that TMC may be considered an interesting target for further development of chemotherapeutic agents against oral cancer.
- Oral cancer
- 3,3’,4’,5’-tetramethoxychalcone
- cell proliferation
- apoptosis
- cell migration
- CCL5/CCR5 axis
- DNA double-strand breaks
Oral squamous cell carcinoma (OSCC) is one of the most common malignant neoplasms in Asian populations (1, 2). It is known that OSCC development is partly facilitated by chronic epithelial irritation and that this tumor occurs more frequently in smokers or individuals who consume excessive amounts of alcohol. Despite advances in diagnostic and therapeutic modalities, the prognosis of OSCC still remains poor, with 5-year survival rates of less than 50%. OSCC exhibits the highest levels of local invasiveness and metastasis to cervical lymph nodes (3). Molecular analyses have shown that the pathogenesis of OSCC is a multi-factorial and multi-step process reflecting the alterations derived from chromosomal instability (4). These events occur via de-regulation of tumor-suppressor genes and the overexpression of numerous oncogenes (5). The outcome of OSCC is mainly dependent on chemotherapeutic drug, radiation, and the feasibility of surgical treatment (5). Non-specific and toxic side-effects in normal cells are commonly observed with the currently available drugs, such as 5-fluorouracil (5-FU), cisplatin and teniposide (6). Currently, there is no effective chemotherapy available for the treatment of unresectable OSCC. Therefore, there is a need to identify potential therapeutic agents that are effective against OSCC and minimize toxicity to surrounding normal cells.
In OSCC, the migration of cancer cells into maxillary and mandibular bones is a common clinical problem (3). Clinical evidence showed high levels of tissue or plasma C-C chemokine ligand 5 (CCL5) [also known as regulated upon activation normal T cell expressed and secreted (RANTES)], which is a marker of an unfavorable outcome in patients with either melanoma, breast, prostate or pancreatic cancer (7). It has been reported that CCL5/C-C chemokine receptor type 5 (CCR5) not only regulates the chemotactic activity in immune cells but also induces cell migration and invasion in several cancer cell types, including OSCC (7). Therefore, inhibition of CCL5–CCR5 signaling might be a novel avenue to control cell migration and invasion of OSCC.
It was reported that synthetic/natural chalcone derivatives have various pharmacological activities such as anti-inflammatory and anti-tumor activities (8, 9). One chalcone derivative, 3,3’,4’,5’-tetramethoxychalcone (TMC), has been reported to inhibit tumor cell proliferation and nitric oxide production (10). However, there has not been a study on TMC addressing its inhibition of OSCC tumor cell proliferation and the underlying molecular mechanism. As part of our study program to identify novel anticancer therapeutic structures (11-17), in this study, a highly invasive human oral cancer cell line, SCC4, was employed to assess the efficacy of TMC as an anticancer agent.
Materials and Methods
Reagents and antibodies. Antibodies against B-cell lymphoma-2 (BCL-2), BCL2-antagonist/killer (BAK), BCL2-associated X protein (BAX), cytochrome c, calpain-1 and calpain-2 were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Antibodies against β-actin, cisplatin, 3-[4,5-dimethyl-2-yl]-2,5-diphenyltetrazoliumbromide (MTT) and 5-FU were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against phosphorylation of checkpoint kinases-2 (p-CHK2), cleaved poly (ADP-ribose) polymerase (PARP) and p-p53 were obtained from Cell Signaling Technology Inc. (Danvers, MA, USA). Antibodies against pro-caspase-3, caspase-3, pro-caspase-9 and caspase-9 were obtained from Biolegend Inc. (San Diego, CA, USA), and antibody against phosphorylation of histone H2AX (γ-H2AX) was purchased from Millipore Inc. (Temecula, CA, USA). Human CCL5 was obtained from PeproTech Inc. (Manchester, UK). A mouse monoclonal antibody that was specific for CCR5 was purchased from R&D Systems (Minneapolis, MN, USA). TMC (Figure 1A) was obtained following the experimental procedures as we reported previously (10).
Cell culture. Human oral cancer cell lines, SCC4, HSC3, and SAS7 (original site of all three: tongue) and human oral fibroblast (OF) cells were kindly provided by Dr. Chih-Hsin Tang (18). The SCC4 cells and HSC3 cells were cultured in Dulbecco's minimum essential medium (DMEM)/F-12 (1:1) (Invitrogen). The SAS7 cells and OF cells were cultured in DMEM. All culture media were supplemented with 10% heat-inactivated fetal bovine serum (HyClone, Logan, UT, USA) and penicillin/streptomycin (Invitrogen). The cells were maintained at 37°C in a humid atmosphere containing 5% CO2.
Cell viability assay. Cells (3×103) were seeded in triplicate on a 96-well plate and treated with different concentrations of TMC (0, 1, 3, and 6 μM) for 24 h. The culture supernatants were then removed and 10 μl of MTT (5 mg/ml) in medium were added to each well. After incubation at 37°C for 2 h, 100 μl of isopropanol was added to dissolve the MTT formazan crystals. The absorbance was determined on a microplate reader at 570 nm (11, 12).
Observation of cell morphology. SCC4 (3×104) cells were cultured in six-well plates for 24 h and treated with different concentrations of TMC (0, 1, 3 and 6 μM) for 24 h. The cell morphology was observed by phase-contrast microscopy (Carl Zeiss, Göttingen, Germany). Images were captured and analyzed by Axiovision software (Carl Zeiss).
Cell-cycle analysis. Cells (3×104) were seeded in six-well plates for 24 h. Briefly, cells were treated with different concentrations of TMC (0, 1, 3 and 6 μM) and vehicle control, dimethyl sulfoxide (DMSO), for 24 h. After treatment, the cells were washed thrice with phosphate buffered saline (PBS), fixed with ice-cold 70% ethanol overnight and stained with 20 μg/ml propidium iodide (Sigma-Aldrich) containing 1 mg/ml RNase (Sigma-Aldrich) and 0.1% Triton X-100 for 1 h. The stained cells were analyzed with a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA). The data were collected using 10,000 cells from each sample and analyzed using Cell Quest software WinMDI (Verity Software House, Topsham, ME, USA). All samples were examined in triplicate in three independent experiments.
Analysis of apoptosis. Apoptotic cell death was detected by terminal deoxynucleotidyl transferase-meditated dUTP-fluorescein nick end-labeling (TUNEL) with fragmented DNA detection kit (Millipore, Temecula, CA, USA) by following the supplier's instruction. Cells (3×104) were seeded in six-well plates and treated with TMC (0, 1, 3, and 6 μM) or vehicle-treated with control. After incubation for 24 h, cells were washed with PBS thrice, fixed in 1% paraformaldehyde for 15 min, and re-suspended in 70% ice-cold ethanol. Cells were permeabilized with 0.1% Triton X-100 and stained with the TUNEL reaction mixture at 37°C for 30 min. The reaction was then blocked in stop/wash buffer for 10 min. The labelled cells were determined using FACScan and the Cellquest program (Becton Dickinson, Lincoln Park, NJ, USA).
Determination of mitochondrial membrane potential. SCC4 cells (3×104) were seeded in six-well plates and treated with TMC (0, 1, 3, and 6 μM) for 24 h. The mitochondrial membrane potential (ΔΨm) was assessed using a fluorometric probe JC-1 (Calbiochem, CA, USA), with a positive charge of a mitochondria-specific fluorophore, indicated by a fluorescence emission shift from green (525 nm) to red (610 nm). After incubation, cells were stained with JC-1 (5 μg/ml) for 30 min at 37°C. Treated samples were analyzed by FACScan using an argon laser (488 nm). Mitochondrial depolarization was specifically indicated by a decrease in the red-to-green fluorescence intensity ratio and analyzed by a FACScan and the Cellquest program (Becton Dickinson, Lincoln Park, NJ, USA).
Western blot analyses. SCC4 cells (1×105) were seeded in 6-cm culture dishes and incubated with or without TMC (0, 1, 3, and 6 μM), or vehicle control, DMSO, for 24 h. Total cell lysates were obtained either from cells that were incubated with TMC or from cells that were incubated with vehicle; the lysates were then boiled in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer for 5 min. The samples were then resolved by 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were incubated at room temperature for 2 h with antibodies against the following: cytochrome c, pro-caspase-3, caspase-3, pro-caspase-9, caspase-9, calpain-1, calpain-2, cleaved-PARP, BCL-2, BAX, BAK, γ-H2AX, p-CHK2, p-p53, β-actin, and CCR5. The membranes were then washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Invitrogen). The proteins of interest were visualized using the ECL western blotting Detection Reagents (GE Healthcare, Piscataway, NJ, USA) and detected using X-ray film (Kodak, Rochester, NY, USA).

Antiproliferative effect of 3,3’,4’,5’-tetramethoxychalcone (TMC) against human oral squamous cell carcinoma cells. A: Chemical structure of TMC. B: SCC4, SAS7, HSC3, and human oral fibroblast (OF) cells were treated with TMC (0-6 μM) and incubated for 24 h followed by cell viability assay.
Immunofluorescence assay. SCC4 cells (3×104) were plated in six-well plates and incubated for 24 h. Cells were incubated with or without TMC (0, 1, 3, and 6 μM). After 24 h, the cells were washed and fixed in 1% paraformaldehyde (Sigma-Aldrich) for 30 min followed by permeabilization with 0.1% Triton X-100 for 30 min. Cells were blocked with 2% bovine serum albumin for 1 h, incubated with γ-H2AX, and probed with Alexa Fluor 594-conjugated anti-rabbit antibody (Invitrogen). The cells were stained with 4’,6-diamidino-2-phenylindole (DAPI) (0.2 μg/ml) for 10 min. Stained cells were visualized using a fluorescence microscope at ×400 magnification.
Migration and invasion assay. The migration assay was performed using Transwell (pore size, 8-mm; Costar, NY, USA) in 24-well dishes. For the invasion assay, filters were pre-coated with 25 μl Matrigel basement membrane matrix (BD Biosciences, Bedford, MA, USA) for 30 min. The experimental protocols were the same for both migration and invasion assays. Cells were pre-treated for 30 min with different concentrations of TMC (1, 3, and 6 μM) or vehicle control (DMSO). Approximately 1×104 cells in 200 μl of serum-free medium were placed in the upper chamber, and 300 μl of the same medium containing 3 ng/ml CCL5 was placed in the lower chamber. The plates were incubated for 24 h at 37°C in 5% CO2. The cells were then fixed in methanol for 15 min and stained with 0.05% crystal violet in PBS for 15 min. Cells on the upper side of the filters were removed with cotton-tipped swabs, and the filters were washed with PBS. Cells on the underside of the filters were examined and counted under a microscope. Each clone was plated in triplicate in each experiment, and each experiment was repeated at least three times. The number of invading cells in each experiment was adjusted by the cell viability assay to correct for proliferation effects of CCL5 treatment (corrected invading cell number=counted invading cell number/percentage of viable cells).
Statistical analysis. Student's t-test was used to calculate the statistical significance of the experimental results between the two groups. A p-value less than 0.05 was considered statistically significant.
Results
TMC inhibited human OSCC cell proliferation. Human oral cancer cells (SCC4, SAS7 and HSC3) and human OF cells were incubated with TMC at different concentrations (0-6 μM) for 24 h. The MTT assay showed that TMC dose-dependently suppressed OSCC cell proliferation (Figure 1B). Interestingly, TMC had no noticeable effect on normal OF cell proliferation (Figure 1B). The IC50 of TMC against SCC4, SAS7 and HSC3 cells were 3, 4.5, and 1 μM, respectively. Noticeably, the tested maximal TMC concentration (6 μM) did not influence on the viability of normal OF cells, indicating its high specificity towards OSCC cells. It was observed that treated OSCC cells exhibited a reduction in cell viability, which differed depending on the cell type and TMC concentrations (Figure 1B). These results indicate that TMC effectively inhibited the proliferation of OSCC cells. SCC4 cells were chosen to investigate the anti-proliferative mechanism of TMC.
TMC induced apoptosis of SCC4 cells. To compare the anti-proliferative effect of TMC with that of 5-FU and cisplatin, SCC4 cells were treated with different concentrations (0-25 μM) of these agents for 24 h, and cell proliferation was determined using the MTT assay. When SCC4 cells were treated with 3 μM TMC, cell proliferation was reduced by approximately 50% compared to 35% and 5% by 5-FU and cisplatin, respectively (Figure 2A). These data indicate that TMC had a superior anti-proliferative effect against SCC4 cells compared with 5-FU and cisplatin (Figure 2A). SCC4 cells were exposed to TMC at 1, 3, and 6 μM for 24 h, and cell morphological observations revealed that a higher number of SCC4 cells were detached, were round in shape and shrunken compared to control and DMSO-treated cells (Figure 2B). We then examined whether TMC could influence the cell-cycle distribution of SCC4 cells. Cells were treated with DMSO or TMC at 0.5, 1, 3, and 6 μM for 24 h, and the cell-cycle phase distribution was analyzed by flow cytometry. As shown in Figure 2C, TMC dose-dependently increased the proportion of SCC4 cells in the sub-G1 phase. Additionally, TMC had a similar cell-cycle inhibitory effect in SAS7 and HSC3 cells (Figure 2D).
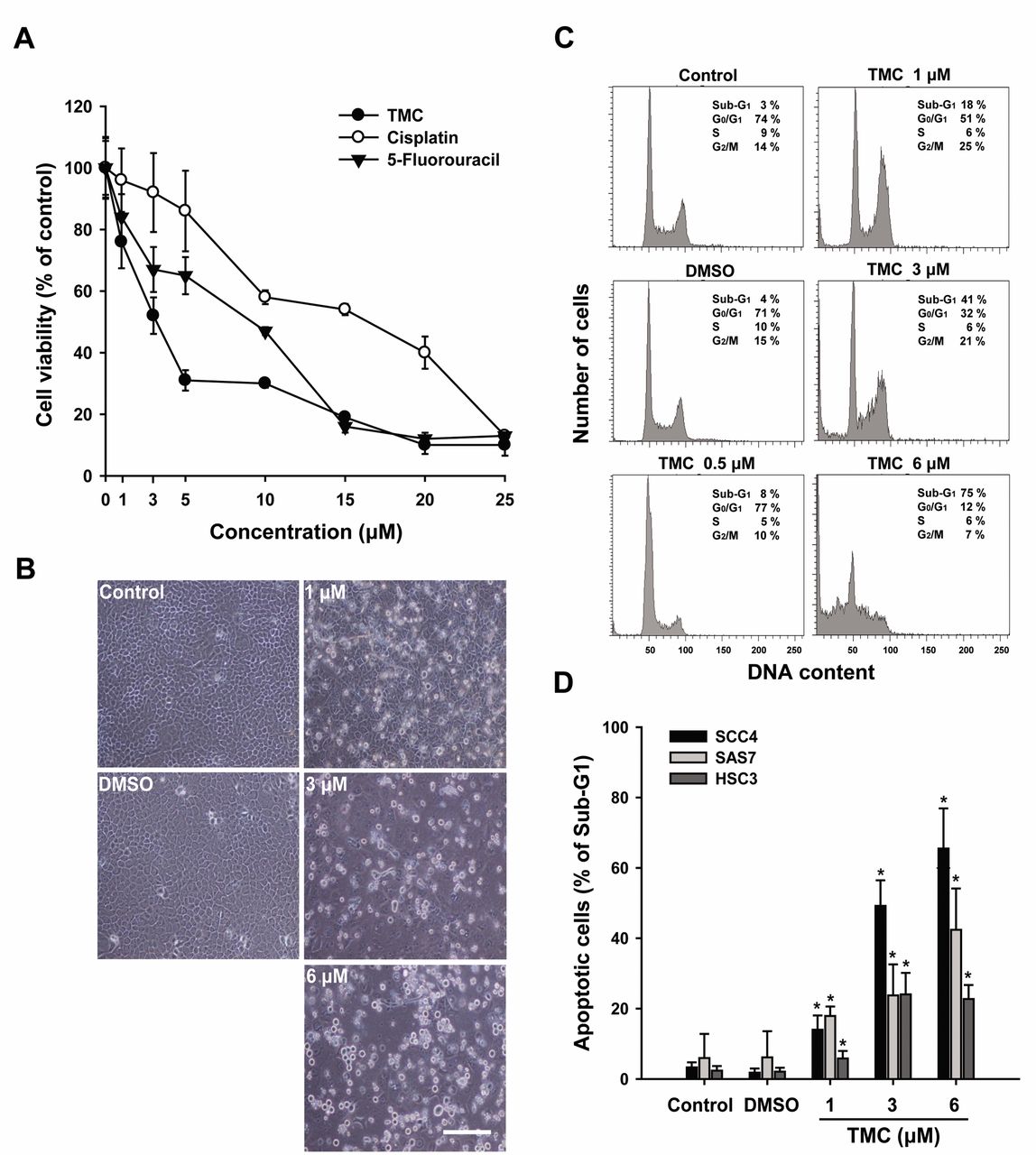
3,3’,4’,5’-tetramethoxychalcone (TMC) inhibits the growth of human OSCC cells. A: Comparing TMC with 5-fluorouracil and cisplatin based on the cytotoxicity against SCC4 cells after 24-h treatment. B: SCC4 cells were incubated with medium-alone (control), vehicle (DMSO), or TMC (1, 3, and 6 μM) and incubated at 37°C for 24 h. The cells were then examined under an inverted optical microscope to assess the effects of TMC. Bars, 50 μm. C: SCC4 cells were incubated at 37°C for 24 h, followed by exposure to medium-alone, DMSO, or TMC (0.5, 1, 3, or 6 μM) for 24 h. The cell-cycle distribution was analyzed by flow cytometry. The percentage of cells in the sub-G1, G0/G1, S, and G2/M phases of the cell cycle are indicated at the right of each histogram. D: The percentage of cells in the sub-G1 phase was calculated and plotted as intensity histograms for the different cell types (SCC4, SAS7, and HSC3 cells). The results are expressed as the mean±standard deviation values for three independent experiments. *p<0.05 was considered statistical significant.
TMC induced mitochondrial dysfunction and up-regulated expression of apoptotic proteins. We further examined the mechanism of cell death induced by TMC. SCC4 cells were treated with TMC for 24 h, and the mitochondrial membrane potential was measured by flow cytometry. It was observed that TMC dose-dependently reduced the mitochondrial membrane potential (Figure 3A). These data demonstrate that TMC influenced mitochondrial function. We then examined whether TMC-induced apoptosis was mediated through the mitochondrial pathway. As shown in Figure 3B, TMC dose-dependently increased the expression levels of apoptosis markers cytochrome c, cleaved caspase-3 and -9, and PARP. Correspondingly, the calcium-dependent family proteins calpain-1 and calpain-2 were also increased in SCC4 cells treated with TMC. Additionally, the effect of TMC on the expression of BCL-2 family proteins was also determined. The results revealed that TMC dose-dependently increased the protein expression of pro-apoptotic BAX and BAK, while reducing that of the anti-apoptotic BCL-2 compared to the control group or cells treated with DMSO (Figure 4A). TMC increased both the BAX/BCL-2 and BAK/BCL-2 ratios (Figure 4B and C). Flow cytometric analysis of TUNEL-positive cells was further employed to detect apoptotic cells. Compared to the control group, the TMC-treated cells contained a higher proportion of TUNEL-positive cells (Figure 4D).
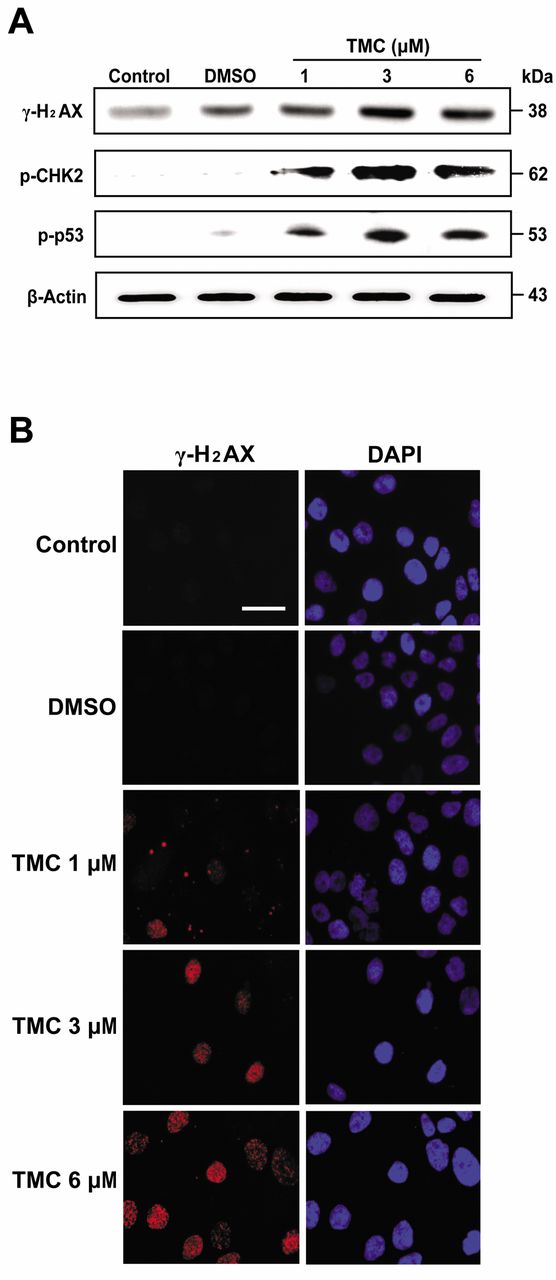
TMC induced DNA double-strand breaks (DSBs) in SCC4 cells. It is known that DNA damage induced by DSBs is followed by the phosphorylation of γ-H2AX (19). Therefore, γ-H2AX is recognized as a DNA damage marker in the first step of recruiting and localizing DNA repair proteins (20). We, thus, determined whether DSBs-related proteins were involved in TMC-induced SCC4 cell apoptosis. As shown in Figure 5A, TMC dose-dependently increased the phosphorylation of γ-H2AX, CHK2, and p53 compared to control or DMSO treatment. We then examined whether TMC induced the formation of γ-H2AX foci after formation of DSBs based on an immunofluorescence assay. Our data showed that TMC dose-dependently induced the γ-H2AX foci formation in the SCC4 cell nuclei (Figure 5B, and C). Taken together, these data demonstrate that TMC-induced apoptosis in SCC4 cells was mediated via the phosphorylation of γ-H2AX, which was triggered by DNA breakage.
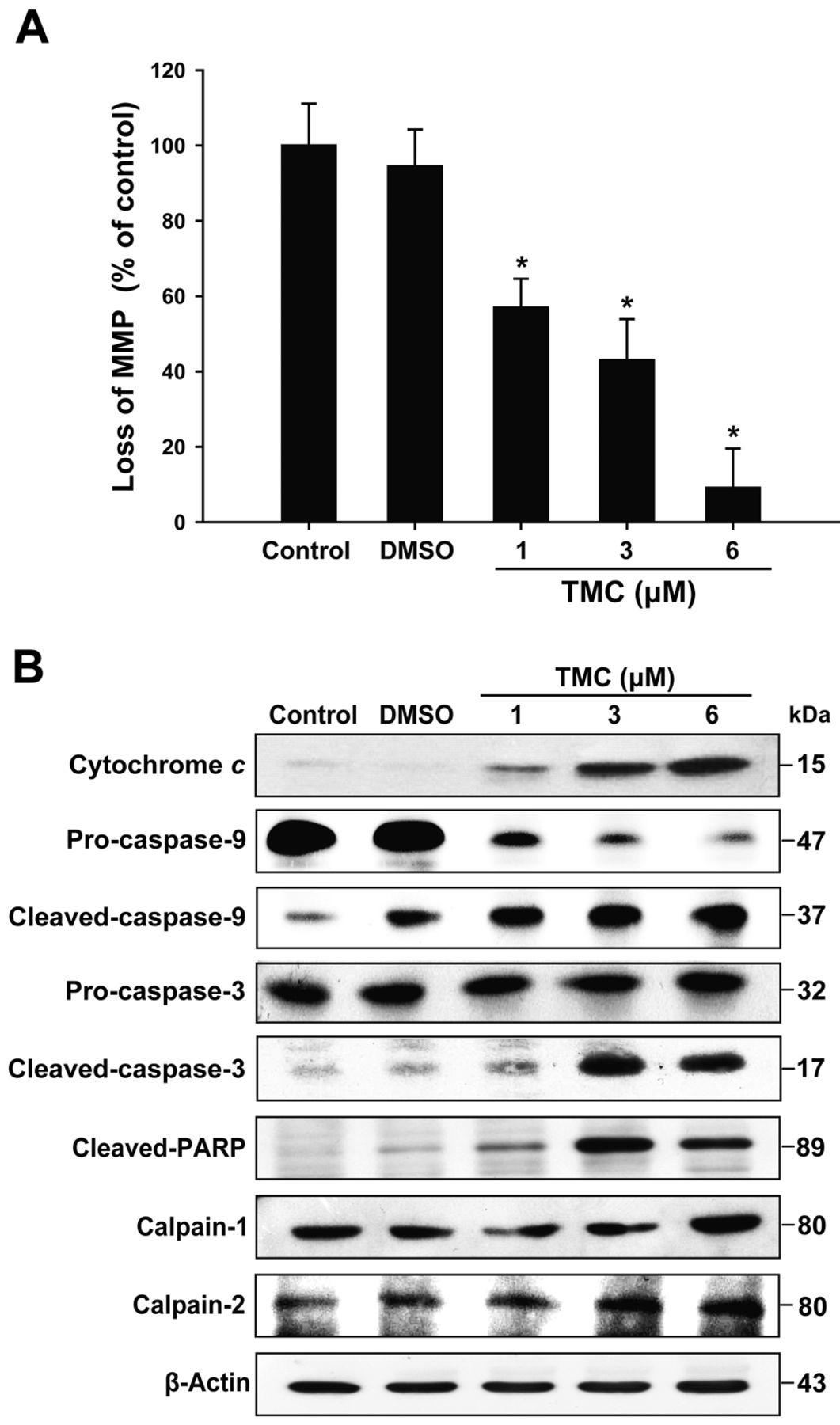
3,3’,4’,5’-tetramethoxychalcone (TMC) induced cell apoptosis through a mitochondria-dependent pathway in SCC4 cells. A: SCC4 cells were treated with different concentrations of TMC at 37°C for 24 h, and the mitochondrial membrane potential (MMP) was examined by a flow cytometric analysis using a fluorometric probe JC-1 as a mitochondrial fluorescent dye. B: The protein expression levels of cytochrome c, caspase-9, caspase-3, calpain-1, calpain-2, and cleaved-poly (ADP-ribose) polymerase (PARP) in cells were examined by western blot. The results are represented for three independent experiments. The statistical significance of the difference was analyzed using Student's t-test (*p<0.05).

3,3’,4’,5’-tetramethoxychalcone (TMC) increased the BCL2-associated X protein (BAX)/B-cell lymphoma-2 (BCL-2) and BCL2-antagonist/killer (BAK)/BCL-2 ratios, and terminal deoxynucleotidyl transferase-meditated dUTP-fluorescein nick end-labeling (TUNEL)-positive staining in SCC4 cells. A: SCC4 cells were cultured at 37°C for 24 h. Cells were incubated with different concentrations of TMC for 24 h, and the expression levels of BCL-2, BAX, BAK, and β-actin were determined by western blot analysis. B and C: The histogram represents both BAX/BCL-2 and BAK/BCL-2 ratios were quantified by using densitometry. D: TUNEL-positive cells were assessed by flow cytometric analysis. The percentage of TUNEL-positive cells was calculated and plotted as intensity histograms. The results are expressed as the mean±standard deviation values for three independent experiments. *p<0.05 was considered statistical significant.
TMC inhibited CCL5–CCR5-directed migration in SCC4 cells. It is known that stimulation with CCL5 directly increases cell migration and invasion via CCR5 receptor of OSCC cells (5). We next examined the inhibitory potential of TMC on SCC4 cell migration. As shown in Figure 6A, CCL5 at 3 ng/ml time-dependently enhanced the migration of SCC4 cells; however, 1 μM of TMC inhibited such enhancement by attenuating the CCL5–CCR5 axis (Figure 6B). Western blot analysis also showed that pre-incubation with TMC (1 μM) significantly reduced CCL5-mediated CCR5 expression (Figure 6B). The results from the present study demonstrate that TMC has the potential to inhibit migration signaled by CCL5–CCR5 axis in SCC4 cells.
Discussion
The ability of cancer cells to evade apoptosis is one of the general characteristics that result in a considerable increase in cancer cell survival (21). Currently, an increasing number of pharmacological agents are derived from synthetic/natural sources. Among these, chalcone derivatives exhibit high cytotoxicity against different types of cancer cells (8). Our present study also demonstrates that a chalcone derivative, TMC, has anticancer activity towards OSCC cells without being cytotoxic to normal cells. It was reported that chalcone analogs inhibit tumor growth of liver cancer (Hep G2) and colon cancer (Colo205) cells (10). However, no study has addressed the precise molecular mechanism of TMC against OSCC. In the present study, the cell viability assay indicated that antiproliferative action of TMC was more potent in OSCC (SCC4, SAS, and HSC3) than in normal cells (Figure 1B). It was reported that in human oral cancer cells, both excision repair cross-complementation group 1 (ERCC1) and the mammalian target of rapamycin (mTOR) are selectively up-regulated resulting in cellular resistance to treatment with cisplatin and 5-FU (22). In the present study, it was found that TMC showed superior cytotoxicity against OSCC cells compared to cisplatin and 5-FU (Figure 2A), which indicates that TMC may have potential as a therapeutic agent for OSCC.


3,3’,4’,5’-tetramethoxychalcone (TMC)-induced formation of DNA double-strand breaks (DSBs) was involved in SCC4 apoptosis. A: SCC4 cells were incubated with TMC for 24 h, and the level of each phospho-protein was assessed by western blot analysis. B: Effect of TMC on phosphorylation of histone H2AX (γ-H2AX) expression in SCC4 cells was detected by immunostaining for γ-H2AX and stained with 4’,6-diamidino-2-phenylindole (DAPI) for nuclear DNA, and then analyzed by fluorescence microscopy. Bars, 50 μm. C: The histogram represents the percentage of cells positive for γ-H2AX foci. The statistical significance of the difference was analyzed using Student's t-test (*p<0.05).
Our previous studies indicate that chalcone derivatives such as 2’-hydroxy-2,3,4’,6’-tetramethoxychalcone cause G1 cell-cycle arrest by p53 activation and inhibit telomerase activity through human telomerase reverse transcriptase inactivation in human lung adenocarcinoma cells (13, 14). The tumor suppressor gene, p53, is a major target for multiple pathways in human cells, including those activated by DNA damage and cellular stress. Formation of DSBs leads to phosphorylation of p53, which is able to suppress tumor growth and enhance therapeutic sensitivity (23). In the present study, we found that TMC induced not only DSBs (Figure 5) but also apoptosis through the p53-mitochondria-dependent apoptotic pathway (Figures 3 and 4). Additionally, treatment of OSCC with TMC affects the BCL-2 family of proteins including activation of BAX and BAK, and suppression of BCL-2 (Figure 4). These results indicated that the effect of TMC against OSCC cell growth is mediated through phosphorylation of p53 and γ-H2AX following the generation of DSBs, all of which activate the mitochondria-mediated apoptotic pathway.

TMC inhibits cell migration in C-C chemokine ligand 5 (CCL5)-treated SCC4 cells. A: SCC4 cells were incubated in a Transwell plate and treated with CCL5 (3 ng/ml) for 12, 24, 36, and 48 h, respectively. Cell migration was analyzed by using the Transwell migration assay. B: SCC4 cells were pre-treated with TMC (1 μM) for 30 min, followed by stimulation with CCL5 for 24 h, and then subjected to an in vitro migration assay. The protein expression level of CCR5 in the cell lysates was measured by western blot. The results are expressed as the mean±standard deviation values for three independent experiments. *p<0.05 was considered to indicate statistical significance.
CCL5 has been demonstrated as an important chemokine that has multiple functions including enhancement of cancer cell migration, invasion, and activation of nuclear factor kappa B through interaction with its specific receptor CCR (24). Increasing CCL5–CCR5 interaction induces migration and invasion by induction of matrix metallopeptidase-9 expression in human OSCC, particularly in SCC4 cells (18). In the present study, we found that CCL5 increased the migration of SCC4 cells (Figure 6A). However, the CCL5-stimulated OSCC migration was attenuated by TMC. In addition, TMC treatment reduced CCL5-mediated CCR5 expression (Figure 6B). Taken together, these results provide evidence that TMC has a dual function in OSCC cells by the induction of cell apoptosis and the inhibition of cancer cell migration.
In conclusion, this study demonstrated that TMC is a potent inhibitor of OSCC cell proliferation. TMC induced both DSBs and the p53-mitochondria-dependent apoptotic pathway in OSCC cells. Moreover, TMC reduced OSCC migration by attenuating the CCL5–CCR5 axis. The present study may be useful in developing TMC as a therapeutic agent for treatment of refractory oral cancer.
Acknowledgements
This work was supported by the National Science Council, Taiwan (NSC 100-2113-M-324-001-MY3, NSC101-2313-B-039-004-MY3 and NSC 101-2811-M-324-001), China Medical University, Taiwan (CMU102-S-28), and the Tomorrow Medicine Foundation. Experiments and data analysis were performed through the use of the microscopic facility at Scientific Instrument Center of Academia Sinica and with the assistance of Dr. Shu-Chen Shen.
Footnotes
-
↵* These Authors contributed equally to this work.
-
Conflicts of Interest
The Authors declare that there are no potential conflicts of interest.
- Received December 28, 2013.
- Revision received January 24, 2014.
- Accepted January 27, 2014.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.