


Abstract
Aim: We investigated the efficacy and safety of capecitabine and gemcitabin (GemCap) in heavily pre-treated, therapy-resistant metastatic colorectal cancer (mCRC) patients and the clinical importance of cell-free DNA (cfDNA) measurement. Patients and Methods: Patients' inclusion criteria included histopathologically-verified mCRC refractory to standard chemotherapy, adequate organ function and performance status. Treatment included capecitabine (2,000 mg/m2 day on days 1-7 q2w) and gemcitabine (1,000 mg/m2 on day 1). The number of DNA alleles was measured in pre-treatment plasma samples using an in-house developed qPCR. Results: Forty-nine patients were included in the study. GemCap was well-tolerated in the majority of patients. Disease control rate was 30%, median progression-free survival (PFS) and overall survival (OS) by intention-to-treat were 2.7 (95%CI=2.6-2.8) and 6.8 (95%CI=5.0-7.7) months. Median OS in patients with cfDNA concentrations above the median (13,200 alleles/ml) was 4.7 (3.7-9.6) months compared to 7.8 months in the remaining patients (HR=2.22; 1.07-3.9; p=0.0186). The prognostic value of the cell-free DNA (cfDNA) was confirmed by multivariate analysis. Conclusion: GemCap was well-tolerated with encouraging efficacy, and cfDNA was shown to hold a strong prognostic value.
A large fraction of patients with metastatic colorectal cancer show primary resistance to available treatment options and even those primary sensitive to standard agents eventually develop resistant disease. Treatment of refractory disease is, therefore, a major challenge. At this stage, some patients will be in good performance status and can benefit from yet another treatment option with the aim of stabilising disease, prolonging the time-to-progression, and improving the quality of life and overall survival time.
Gemcitabine has shown efficacy against several solid tumours but seems to show minimal activity in mCRC as a single-agent, and is not standard for this cancer type (2). However, there is strong pre-clinical evidence of a synergistic effect of CRC cell lines with fluoropyrimidines in vitro. Gemcitabin acts synergistically with fluoropyrimidines to enhance binding of thymidylate synthase and increase the inhibition of DNA synthesis (3-4), and different combinations have confirmed anti-tumor activity in patients with gastrointestinal solid tumors, including colorectal cancer (5). In addition, these regimens in general have a manageable low-toxicity profile. Results of 8 phase-II trials are summarised by Merl et al. who concluded that the combination showed response rates of 30-38% and favourable TTP and OS between 4-83 months and 9.8-18 mo, respectively (5). Anti-tumour activity was also observed in heavily pre-treated patients, which suggests that these combinations may be potential feasible options for patients with chemotherapy-refractory mCRC, who have failed to respond to all standard therapies (…). Capecitabine is an orally-administered pro-drug to 5-fluorouacil, which is convenient for treatment in this setting.
Translational research could help improve patient selection for relevant therapy and to avoid treatment with no benefit and unnecessary harmful toxicity. We have previously investigated circulating cfDNA measured in the peripheral blood of patients with heavily pre-treated mCRC. The levels of cfDNA alleles were observed to hold a strong prognostic value and high levels prior to therapy were consistently associated with a poor survival in two different phase II studies (6-8). The prospective collection of blood samples for TR studies was therefore also a focus of this study.
The present study aimed to investigate the efficacy and safety of the combination of capecitabine and gemcitabin (GemCap) in heavily pre-treated, therapy-resistant mCRC patients and to validate previous observations of the clinical importance of cell-free DNA measurements in similar patient cohorts.
Patients and Methods
Study design. The study was a single-arm phase II study and the sample size was calculated according to Simon's two-stage design (9). The Simon's two-stage minimax design ensures termination of the study in case of a minimal effect of the treatment: The sample size calculation was based on the following criteria: a disease control rate of 40% after three months was considered clinically relevant with the perspective of further investigations, whereas less than 20% was deemed unacceptable. Alpha was set to 0.05 and β=0.2, for a power of 80%. Consequently, the first step in the trial was planned to include 18 patients. If 4 or more patients after the first evaluation scan obtained disease stabilisation, the study would be continued into the second step, which would include another 15 patients. The total patient population was, therefore, calculated to be 33 patients. Only patients who had received three months of therapy and were evaluable for response according to RECIST would be included in the efficacy analysis, unless there was unequivocal clinical progression.
The translational research investigation was performed prospectively, with the collection of blood samples prior to the first and each subsequent cycle, as well as at each follow-up visit. Analysis of cfDNA was a pre-defined objective.
Thus, the primary end-point was the rate of patients free from progression at 3 months, and secondary objectives included progression-free survival, overall survival, safety, compliance and the identification of predictive and prognostic markers.
Patient selection. Inclusion criteria included: Patients with histologically-verified mCRC, previously exposed to and refractory to fluoropyrimidines, oxaliplatin and irinotecan, measurable disease according to RECIST version 1.1, age ≥18 years, ECOG PS 0-2, adequate bone marrow, renal and hepatic function and consent to sampling for TR.
Patients who were excluded from the study had clinical signs of or verified brain metastasis or required radiotherapy against target lesions, other clinically-significant concurrent illness (at the discretion of the investigator), were pregnant or breastfeeding, had received other experimental therapy within 30 days of the study entry or had suffered from any other malignant disease within 5 years of inclusion in the study (except basal cell carcinoma of the skin and cervical carcinoma in situ). Concurrent vaccination against yellow fever was also not allowed.
All patients provided signed informed consent before study entry and the protocol was approved by the Danish Medicines Agency and The Regional Scientific Ethical Committee of Southern Denmark. The study was conducted in accordance with the Good Clinical Practice guidelines, as issued by the International Conference on Harmonisation and the Declaration of Helsinki.
Treatment. Patients received capecitabine orally 2,000 mg/m2 on day 1-7 (to a maximum of 4,000 mg daily) and gemcitabine 1,000 mg/m2 (to a maximum of 2,000 mg) day 1 q2w administered intravenously. Anti-emetic support was administered according to local guidelines. Postponement of therapy longer than 4 weeks or intake of less than 50% of the prescribed doses during more than three consecutive cycles led to therapy discontinuation.
Comptuted tomographic (CT) scans of the chest and abdomen were used for evaluation of response according to RECIST version 1.1 and performed at baseline, less than 2 weeks prior to the first cycle and every 12 weeks of therapy during treatment and every three months during follow-up in patients who had stopped treatment without progression. NCI-CTCAE version 4.0 was used for the assessment of toxicity, and was recorded at baseline, prior to each cycle and during clinical follow-up.
Statistics. Progression-free survival (PFS) was calculated from the date of first treatment until date of progression or death from any cause, and overall survival (OS) was defined at time from date of first treatment until death from any cause. The association between marker status and objective response, baseline characteristics and toxicity rates was determined by two-sided t-tests or χ2-test. The Kaplan-Meier method was used to estimate PFS and OS. A multivariate Cox regression analysis was performed to examine whether the different variables were associated with reduced survival. p-Values referred to two-tailed tests and were considered significant when p≤0.05. Statistics were carried out using the NCSS statistical software 2007 v.07.1.5 (NCSS Statistical Software, Utah 84037, USA, www.ncss.com).
Translational research sampling and analysis. After informed consent, pre-treatment blood samples were drawn after inclusion in the study, but prior to the first cycle of therapy. The methods for quantification of cfDNA in the plasma have been described previously (6). Plasma was obtained from blood samples collected in 9 ml EDTA-tubes and centrifuged at 2,000 ×g for 10 min within 2 h and stored at −80°C until use. All samples were analyzed blinded to the study endpoints.
DNA was purified from 1 ml plasma using a QIA symphony virus/bacteria midi-kit on a QIAsymphony robot (Qiagen) according to the manufacturer's instructions. To determine the levels of cfDNA, the amount of the cyclophilin gene (gCYC) was measured using an in-house developed qPCR assay. The in-house assays were based on the Amplification Refractory Mutation System-Quantitative PCR (ARMS-qPCR) methodology and a detailed description has been published recently (6).
Results
Patients and therapy. Fourty-nine patients comprising of 18 females and 31 males, were included in the study. Fifty-five per cent of the patients had been diagnosed with primary, disseminated disease and the median time from primary diagnosis until inclusion in the study was 28 months (range=8-112 months). All patients had progressed on standard cytotoxic therapy; the majority (71%) had also received anti-angiogenesis therapy and 49% had been given anti-EGFR therapy. The pre-treatment characteristics are presented in Table I.
Forty-eight out of the 49 patients commenced treatment as planned and the median number of cycles was 6 (range=0-16). Primary dose reduction of gemcitabine was applied at the discretion of investigator in 2 patients, and of capecitabine in 17 patients due to prior toxicity or frailty of patients. A secondary dose reduction was necessary for gemcitabine in 1 patient and for capecitabine in 17 patients according to clinical guidelines. The reasons for discontinuation were: disease progression in the majority of patients (n=40; 82%), postponed course due to adverse events (n=1; 2%) and at the patient's request (n=5; 10%), n=1 (2%) because of infection, n=1 (2%) due to clinical deterioration and a single patient (2%) did not start therapy. A total of 15 patients prematurely stopped therapy before the 3rd month and were not available for radiological response evaluation. These included 7 individuals who did not have an evaluation scan at the time of discontinuation, and 8 patients who had scans brought forward due to symptoms of disease progression, which was verified. The study was designed to include 18 evaluable patients in the interim analysis and continue onto step 2 if 4 or more patients responded. This criterion was fulfilled and the study was continued; 33 patients were evaluable for response after 3 months of therapy.
Patients' pre-treatment characteristics in GemCap.
Efficacy. Ten patients evaluable for response after three months of treatment reached disease stabilisation (30%) (10/33 patients), but no objective responses were observed. The rate of disease stabilisation as best overall response at any time was calculated as 10/41 (24%). At the time of this analysis, seven patients were still alive, and the median PFS and OS as intention-to-treat analysis are 2.7 (95%CI=2.6-2.8) and 6.8 (95%CI=5.0-7.7) months, respectively.
Safety. All AEs and SAEs were recorded, including disease-related events. There were no treatment-related deaths or SUSARs reported, but 33 SAEs were recorded. Only three of these were possible therapy-related incidents, including three patients with fever without infectious focus. Thus, the majority of AEs reflected the severe disease status and the rapid progressive nature at this stage. We observed no grade 4 AEs during therapy and the 11 grade 3 AEs were documented in 5 patients experiencing grade 3 fever without neutropenia and in 6 who developed grade 3 fatigue (Table II). We recorded 2 cardiovascular events which were not present at the baseline of the study. One episode was observed in a patient previously known to have asymptomatic well-treated atrial fibrillation, who developed chest pain during pneumonia, but acute ischemia was excluded. The other patient had been monitored for chest pain during previous chemotherapy regimens, and ischemia was not observed. The recorded event was a similar short episode of chest pain, but ischemia was excluded and the patient quickly recovered. Both events were recorded as mild and unrelated to therapy. In general, the treatment was well-tolerated.
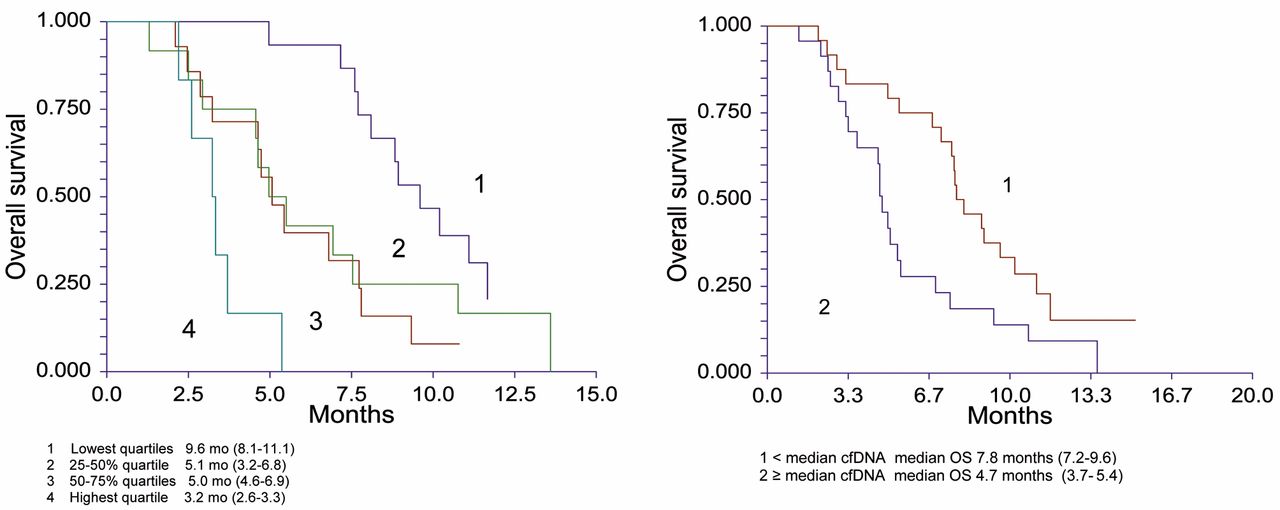
Kaplan-Meier major OS curves according to cfDNA levels in quartiles and patients dichotomised by the median level of alleles per ml.
Baseline and highest symptom grade recorded during therapy.
Translational research. Fourty-seven of the 49 patients had pre-treatment blood samples available for cfDNA measurement. The median cfDNA levels in the total cohort was 13,200 (range=1,000-549,500) alleles/ml. The level of cfDNA was investigated in relation to all pre-treatment characteristics as shown in Table I. There were no significant differences in the levels of cfDNA according to different pre-treatment characteristics.
Kaplan-Meier survival plots revealed a shorter OS with increasing levels of pre-treatment cfDNA when divided into quartiles of cfDNA levels. The median OS in patients with the highest level of cfDNA was 4.7 months (3.7-9.6 months) compared to 7.8 months in the remaining patients, (Hazard ratio (HR)=2.22, 1.07-3.9; p=0.0186) (Figure 1). A multivariate cox analysis including age (more or less than 63 years), PS, gender and cfDNA (divided into quartiles) confirmed the independent prognostic value of cfDNA in this cohort. The HR for each step increase of cfDNA was 1.7 (95% CI=0.3-2.4; p=0.0009).
Discussion
The combination of gemcitabine and capecitabine was tested as a third- or fourth-line therapy for patients with mCRC having failed on all available standard agents, and proven to be feasible with moderate activity in approximately one third of our patients. The study was designed to evaluate the fraction of patients with disease control after 3 months of therapy, and the regimen chosen aimed to prioritise the quality of life by using a low-toxicity schedule and dose reduction to avoid severe toxicity. In general, the treatment was very well-tolerated, as illustrated by the low number of grade 3 events, with the majority being related to disease rather than toxicity.
The unmet need for a treatment option after failure of standard agents has placed a high demand on the design of studies. Defining the patient population clearly (i.e. whether the patients are previously exposed to or truly refractory to therapy) is important. Some trials report on previously used chemotherapy, whereas others have more precise definitions of therapy resistance such as progression within 3 months after the end of chemotherapy, or progression whilst on treatment. These aspects complicate a relevant comparison between studies. Another important aspect is the choice of end-point in these late-stage trials. With the low response rates and poor prognosis in this group of patients, results are highly influenced by the interval between re-stagings. We chose to evaluate patients after 3 months to avoid multiple visits and our data are therefore not comparable to most studies in the literature presenting data on early evaluations i.e. at 6 weeks and 8 weeks (10). Despite these differences, this phase II trial presents PFS and OS data which compare favourably with data presented in the most recent literature.
The rate of disease control was 30% at three months, and the median PFS and OS as intention-to-treat analysis were 2.7 (95%CI=2.6-2.8) and 6.8 (95%CI=5.0-7.7) months, respectively. Only a few studies have investigated gemcitabine and fluoropyrimidines in the third- or fourth-line setting. A small retrospective report by Saif and colleagues used similar treatment regimen to ours (10). Re-staging was performed on an 8-weekly basis and tumor reduction was observed in 4 patients; the DCR was 63.6%. No survival data were reported, but the authors conclude that therapy was feasible and could be considered as an option for these patients. Another study prospectively investigated protracted 5-FU infusion and gemcitabine in 4-weekly schedules and used a similar end-point evaluation of 3 monthly scans. The disease control rate was 62.2%, median TTP 4.2 and median OS 8.9 months. Results were found to be encouraging (11). In the review summarised by Merl et al., it was concluded that gemcitabine and 5-FU could provide a feasible palliative therapy for refractory mCRC as long as a bolus of 5-FU was not used (5). However, the number of studies addressing true refractory patients is still limited. The number of phase II trials investigating new options has increased during the last decade and include agents like MMC, vorinostat, S1, capecitabine and bevacizumab (12-15). In general, tumour reduction is rarely observed and the median PFS and OS are comparable with the ones of the present study. However, no regimen has stood out as an obvious choice for larger trials, and a true effective therapy for heavily pre-treated mCRC is yet to be identified.
Finding the right balance between a possible efficacy and quality of life has proven difficult in this setting, and randomised studies will, therefore, be of high value. Only a few randomised trials have been conducted in refractory disease. The results of the recently published CORRECT trial investigating the small molecule TKI regorafinib against placebo also illustrated the rapid progressive nature of the disease (16). The study demonstrated a modest benefit from the active drug, in terms of rare tumour reduction (1%), and an increase in median OS. Median overall survival was 6.4 months in the regorafinib group and 5.0 months in the placebo group, and the HR was 0.77 (95% CI=0.64–0.94; p=0.0052). The DCR at 6 weeks was 41% in the regorafinib arm and 15% in the placebo arm and the median PFS were 1.9 and 1.7 months, respectively.
With the poor prognosis in this setting, predictive and prognostic markers for outcome are important. Advances in technology has enabled us to develop a feasible assay for the quantification of cfDNA alleles in the peripheral blood (6). In three previous phase II studies we investigated the concentration of cfDNA in plasma from patients with heavily pre-treated mCRC, and found impaired survival with increasing levels at baseline. Although the importance of cfDNA in cancer has recently attracted great focus (17), there are no studies presented in the literature from similar settings. The first study we presented included patients treated with third-line cetuximab and irinotecan (6), the second included temsirolimus and irinotecan (7) and the third used pemetrexed and gemcitabine (8). Despite the small sample size of the present study, data presented here support our previous observations. The median OS was significantly higher in patients with low levels of cfDNA (HR=2.22; p=0.0186) which suggests that levels of cfDNA reflect disease biology and could become an important marker for outcome. The predictive value of a marker cannot be reliably investigated in non-randomised phase II trials, but cfDNA is clearly a good candidate for biomarker studies in future randomised trials in CRC.
In conclusion, the present phase II study supports the literature, which suggests that GemCap could be a feasible palliative treatment for refractory mCRC; however, future trials should be designed to address both quality of life, improvement of outcome and better selection for therapy. Randomised trials would aid in these aspects and cfDNA measurements are a promising candidate for biomarker studies in this setting.
Acknowledgements
We sincerely thank the Research counsil, Lillebaelt Hospital and Asta og Peter Gøtz-Petersens Fundation for funding of the study.
Footnotes
-
↵* Current affiliation: Department of Oncology, Aarhus University Hospital, DK. Danish Colorectal Cancer Group South,Vejle Hospital, Kabbeltoft 25, 7100 Vejle, Denmark.
-
Conflicts of Interest
The Authors declare no conflicts of interest/have no disclosures.
- Received September 21, 2013.
- Revision received December 16, 2013.
- Accepted December 17, 2013.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}