


Abstract
Currently used platinum drugs fail to provide long-term cure for ovarian cancer mainly because of acquired drug resistance. In this study, a new monofunctional planaramineplatinum(II) complex, namely tris(8-hydroxyquinoline)monochloroplatinum(II) chloride (coded as LH3), was synthesised and investigated for its activity against human ovarian A2780, cisplatin-resistant A2780 (A2780cisR) and ZD0473-resistant A2780 (A2780ZD0473R) cancer cell lines, alone and in combination with the phytochemicals curcumin, genistein and resveratrol. Cellular levels of glutathione in A2780 and A2780cisR cell lines before and after treatment with LH3 and its combinations with genistein and curcumin were also determined. Interaction of the compounds with salmon sperm DNA, pBR322 plasmid DNA and damage to DNA in A2780 and A2780cisR cells due to interaction with LH3-alone and in combination with phytochemicals were also investigated. LH3 was found to be much more active than cisplatin against the resistant tumor models and greatest synergism in activity was observed when combinations of LH3 with genistein and curcumin were administered as a bolus. For combinations of LH3 with the phytochemicals, platinum accumulation and the level of Pt–DNA binding were found to be greater in the resistant A2780cisR cell line than in the parental A2780 cell line. Greater activity of LH3 than cisplatin against the resistant ovarian cell lines indicates that it may have the potential for development as a novel anticancer drug and that its combination with phytochemicals can serve to further enhance drug efficacy.
Ovarian cancer remains the leading cause of death from gynaecological types of cancer in women in the Western world mainly due to the development of drug resistance and late detection (1). Although tumor generally responds well to the combination of platinum with paclitaxel, the drugs fail to function when relapse often occurs (1). As a part of our continued search to find new tumour-active platinum compounds (2, 3), herein we report the synthesis, characterization, and activity against human ovarian A2780, A2780cisR and A2780ZD0473R cancer cell lines of tris(8-hydroxyquinoline)monochloroplatinum(II)chloride (coded as LH3). The interactions of the compound with salmon sperm DNA, pBR322 plasmid DNAs and damage to cellular DNA were also investigated.
With the idea that phytochemicals can sensitize tumor cells towards apoptotic action by chemotherapeutics due to modulation of various cellular pathways involved in chemoresistance (4, 5), we also determined cell kill due to combinations of LH3 with phytochemicals genistein, curcumin and resveratrol. Genistein (Figure 1) is a soy bean-derived isoflavone that acts as an inhibitor of protein tyrosine kinase, thus attenuating the growth of cancer cells (5-7). It can suppress the tumor necrosis factor (TNF)-induced activation of NF-κB, degradation of Kappa-Bungarotoxin (1κBα), nuclear translocation of p65 and subsequent gene expression. It can also inhibit the protein kinase B (AKT) pathway, thus enhancing necrotic-like cancer cell death (8). Recent studies have shown that genistein can suppress growth of gynaecological carcinomas (9). Curcumin (Figure 1) is a key component of turmeric that has antioxidant, antitoxic, anti-inflammatory, cancer-preventive and potentially chemotherapeutic properties (10, 11). It causes cancer cell death via multiple targets including pro-apoptotic proteins p53 and Bcl-2-associated X protein (BAX), transcription factors NF-κB, AKT and p38 mitogen-activated protein kinase (MAPK), and growth factors such as epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) (12-15). Resveratrol (Figure 1), present in red grapes and mulberries, has both chemopreventive and chemotherapeutic potential (10, 16). In combination with chemotherapeutics, resveratrol can both enhance and reduce cancer cell death. For example, resveratrol has adverse effects when used in combination with paclitaxel, vincristine and daunorubicin in some tumour cells by inducing S phase cell arrest, thus preventing cells from entering into the G2/M phase, where paclitaxel exerts its effect (17-19).

Structures of LH3, cisplatin, curcumin, genistein and resveratrol.
In the present study, we investigated the effect of presence of curcumin, genistein and resveratrol on the efficacy of LH3. Changes in cellular glutathione levels in A2780 and cisplatin-resistant A2780cisR ovarian cancer cells were also determined before and after their treatment with LH3 alone and its sequenced combinations with genistein and curcumin.
Materials and Methods
Materials. Potassium tetrachloroplatinate (II) [K2 (PtCl4)], N,N-dimethylformamide (DMF) (C3H7NO) and 8-hydroxyquinoline (C9H7NO) were from Sigma Chemical Company (St. Louis, MO USA) HCl was from Ajax chemicals (Auburn NSW, Australia); ethanol was from Merck Pty Ltd. (Kilsyth, Australia).
Synthesis. K2PtCl4 (0.5 mmol, 0.210 g) was dissolved in milli-Q (mQ) water (10 ml) and treated with concentrated hydrochloric acid (0.25 ml). The temperature of the solution was increased to 50°C followed by the addition of 8-hydroxyquinoline (5 mmol, 1.4516 g) dropwise over 1 h. The mixture, protected from light, was stirred at room temperature for two weeks followed by the addition of 20 ml of 0.25 M hydrochloric acid. Stirring was continued for one week at room temperature in the dark. The mixture was centrifuged at 5,500 rpm for 10 min to collect precipitate of LH3. The crude product was washed with ice-cold ethanol and purified by dissolving in 0.05 M HCl. The yield of the product was 68% (242 mg) as a light orange powder.

Numbering scheme for 8-hydroxyquinoline.
Characterization. C, H, and N were determined using a Carlo Erba 1106 automatic analyzer available at the Australian National University ACT Australia. Pt was determined by graphite furnace atomic absorption spectroscopy (AAS). As LH3, could not be obtained in crystalline form, infrared (IR), mass (MS) and 1H nuclear magnetic resonance (NMR) spectra were used to aid in structural characterization of the compound. The IR spectrum was obtained using a Cary 660 FT-IR spectrometer. (Agilent Technologies, Mullgrave, Victoria, Australia). To obtain the MS spectrum, a solution of LH3 made in 90% methanol and 10% DMF was sprayed into Finnigan LCQ mass spectrometer available at the School of Chemistry The University of Sydney Australia. To obtain the 1H NMR spectrum, the compound was dissolved in deuterated dimethyl sulfoxide and prepared in a 5 mm high-precision Wilmad NMR tube. A Bruker DPX400 spectrometer (Bruker Pty LTD, Preston, Victoria, Australia) was used with frequency of 400.2 MHz. In 1H NMR, s, d and q denote singlet, doublet and quartet, respectively. The numbering scheme adopted for 8-hydroxyquinoline is given in Figure 2.
Light orange powder (214 mg, 60%); 1H NMR DMSO δ ppm: 8.82 (s, due to OH); 8.6 (d, due to C2H); 7.7 (d, due to C4H); 7.4 (q, due to C9H); 7.0 (q, due to C3H); 3.5 (s, due to water); 2.5 (s, due to DMSO); IR (KBr): 3279, 3056, 23609, 2341, 1574,1501,1459,1375,1281, 1114, 824,740, 661 cm−1; MS (ESI) m/z (%): 483.05 (100)=[Pt(C9H7NO)3Cl2 − (C9H7NO)− 2Cl − H]; 482.05 (83)=[Pt(C9H7NO)3Cl2 − (C9H7NO) − 2Cl −2H]; 434 (70) =[Pt(C9H7NO)3Cl2 − (C9H7NO)2 + H2O + 4H]; 506 (82)= [Pt(C9H7NO)3Cl2 − (C9H7NO) − Cl − H2O + 3H]; 507 (70)=[Pt(C9H7NO)3Cl2 − (C9H7NO) − Cl − H2O + 4H]; 505 (65)=[Pt(C9H7NO)3Cl2 − (C9H7NO) − Cl − H2O + 2H]; 973.12 (80)=[Pt(C9H7NO)3Cl2 + (C9H7NO)2 − H2O]; 974.15 (70)=[Pt(C9H7NO)3Cl2 + (C9H7NO)3 − H2O + H]; 972.15 (60)=[Pt(C9H7NO)3Cl2 + (C9H7NO)3 − H2O − H]; elemental analysis (anal. calcd) for C27H21Cl2N3O3Pt: C 46.3, H 3.0, N 5.9, Pt 28., found: C 46.3±0.4, H 2.9±0.4, N 5.9±0.4, Pt 28.2±1.0).
Molar conductivity. Molar conductivity values of solutions of LH3 and cisplatin were determined at concentrations ranging from 60 μM to 200 μM. Firstly, to obtain 1 mM solutions, compounds were dissolved in 1:4 mixture of DMF and mQ water in the cisplatin and 2:1 mixture of DMSO and mQ water in the case of LH3. The limiting molar conductivity values at zero concentration (Λo) were obtained from molar conductivity versus concentration plots (not shown).
Biological Activity
Interaction with DNA. Interaction of LH3 and cisplatin (used as a reference compound) with salmon sperm DNA (ssDNA) and pBR322 plasmid DNA (without and with BamH1 digestion) was investigated using agarose gel electrophoresis. The amount of DNA was kept constant while the concentrations of the compounds were varied.
ssDNA. Stock solution of ssDNA having concentration ranging from 1 mg ml−1 to 1.5 mg ml−1 was prepared by dissolving 10-15 mg of ssDNA in 10 ml of 0.05 M Trizma buffer at pH 8 and it was stored at −18°C until used. Aliquots of ssDNA (4 μl at 1.5 mg ml−1) were added to solutions of varying amounts of LH3 and cisplatin and the total volume was made up to 20 μl by adding mQ water so that the concentrations of compounds ranged from 1.87 to 60 μM. of mQ water (16 μl) was added with 4 μl of ssDNA to provide a DNA blank. Incubation was carried out in shaking water-bath at 37°C for 2 h, following which 16 μl aliquots of drug–DNA mixtures were loaded onto the 2% agarose gel and electrophoresis was run in tris-acetate-EDTA (TAE) buffer for 50 min at 120 V cm−1 at room temperature.
pBR322 plasmid DNA. pBR322 plasmid DNA was supplied in a 10 mM Tris-HCl, pH 7.5, 1 mM EDTA buffer at a concentration of 0.5 μg/μl. Exactly 1 μl of supplied pBR322 plasmid DNA in solution was added to varied amounts of solutions of LH3 and cisplatin at different concentrations ranging from: 1.87 to 60 μM. The total volume was made up to 20 μl by adding mQ water. The DNA blank was prepared by adding 19 μl of mQ water to 1 μl of pBR322 plasmid DNA. The samples were incubated for 5 h on a shaking water-bath at 37°C in the dark, at the end of which the reaction was quenched by rapid cooling to 0°C for 30 min. The samples were thawed and mixed with 2 μl of marker dye (0.25% bromophenol blue and 40% of sucrose). Aliquots of drug–DNA mixtures (17 μl) containing 1 μl of DNA were loaded onto the 2% agarose gel made in TAE buffer that contained ethidium bromide (1 mg ml−1). At the end of the electrophoresis, the gel was stained in the same TAE buffer (20) at room temperature for 1 h and 30 min at 150 V cm−1.
BamH1 digestion. In this series of experiment, an identical set of drug–DNA mixtures, as that described previously, was first incubated for 5 h in a shaking water-bath at 37°C and then subjected to BamH1 (10 units μl−1) digestion. To each 20 μl of incubated drug–DNA mixture were added 3 μl of 10× digestion buffer SB followed by 0.2 μl BamH1 (2 units). The mixtures were left in a shaking water bath at 37°C for 1 h, at the end of which the reaction was terminated by rapid cooling. Electrophoresis was carried out and the gel was subsequently stained with ethidium bromide, visualized under short UV light and photographed using a Kodak Gel Logic 100 imaging system (GL 100) available in the Discipline of Biomedical Science The University of Sydney and images were analysed using Kodak MI molecular imaging software.
Cytotoxicity assay. The cell kill due to drugs was determined using 3-(4,5-di-methyl-2-thiazole)-2,5-diphenyl-2H-tetrazolium bromide (MTT) reduction assay (25, 26). Briefly, 4,000 to 5,500 cells (maintained in logarithmic growth phase in complete medium consisting of RPMI-1640, 10% heat-inactivated fetal bovine serum (FBS), 20 mM HEPES, 0.112% sodium bicarbonate, and 2 mM glutamine without antibiotics) were seeded into flat-bottomed 96-well culture plates in 10% FBS/RPMI-1640 culture medium and allowed to attach overnight. For single treatments, drugs were administered at three to five different concentrations ranging from 0.16 to 20 μM for cisplatin and LH3, and 0.16 to 200 μM for genistein, resveratrol and curcumin, 100 μl of drugs were added to equal volumes of cell culture in triplicate wells, then left to incubate under normal growth conditions for 72 h at 37°C in a humidified atmosphere (containing 5% carbon dioxide in air). For combination studies, increasing concentrations of drugs at equipotent ratios were administered to the cells using the sequences: 0/0 h. 0/4 h and 4/0 h, where 0/0 h meant that both the drugs were administered at the same time, 0/4 h meant that LH3 was administered first followed by that of the phytochemical (genistein/curcumin/resveratrol) or cisplatin 4 h later, and 4/0 h meant the converse. At the completion of the 72 h incubation period, the MTT assay was performed.
Median-effect analysis was carried out to calculate combination index (CI) as a quantitative measure of combined drug action. This was based on the pooled data from three to five individual experiments each comprising at least three data points for each drug alone and for each drug combination. CI for two drugs in combination was calculated using the following equation (21, 22) and using Calcusyn software (V2) (Biosoft, Cambridge, UK):
 where D1 and D2 are the concentrations of compounds 1 and 2 in combination needed to achieve x% inhibition, whereas D1x and D2x represent the same when they are present alone. In the following equation, Dx denotes dose of drug, Dm is the median-effect dose which is equivalent to the drug concentration required for 50% cell kill (IC50), fa is the fraction of cells affected (i.e. killed) by the dose, fu is the fraction of cells remaining unaffected so that fu=1−fa and m is the exponent defining the shape of the dose–effect curve.
where D1 and D2 are the concentrations of compounds 1 and 2 in combination needed to achieve x% inhibition, whereas D1x and D2x represent the same when they are present alone. In the following equation, Dx denotes dose of drug, Dm is the median-effect dose which is equivalent to the drug concentration required for 50% cell kill (IC50), fa is the fraction of cells affected (i.e. killed) by the dose, fu is the fraction of cells remaining unaffected so that fu=1−fa and m is the exponent defining the shape of the dose–effect curve.
 CI of <1, =1 and >1 indicate synergism, additivity and antagonism, respectively, in the combined drug action. The linear correlation coefficient, r, was used as a measure of goodness of fit for the pooled data (where r=1 is a perfect fit). For the cell culture system r should be greater than 0.95.
CI of <1, =1 and >1 indicate synergism, additivity and antagonism, respectively, in the combined drug action. The linear correlation coefficient, r, was used as a measure of goodness of fit for the pooled data (where r=1 is a perfect fit). For the cell culture system r should be greater than 0.95.
Platinum accumulation and Pt–DNA binding. Cellular accumulation of platinum and the level of platinum-DNA binding were determined for most synergistic (0/0 h) and most antagonistic (4/0 h) combinations of LH3 with genistein, curcumin, resveratrol and cisplatin. The aim was to determine whether the values were affected by the sequence of administration or whether there was any correlation between the combined drug action and the level of Pt–DNA binding. The method used for the determination of total intracellular platinum and platinum–DNA level was a modification of that described by Di Blasi et al. (23). Compounds were added to culture plates containing exponentially growing A2780 and A2780cisR cells in 5 ml 10% FCS/RPMI-1640 culture medium (cell density=1×106 cells ml-1). Incubation was carried out for 24 h at the end of which cell monolayers were trypsinized and cell suspension (5 ml) was transferred to centrifuge tube and spun at 3500 rpm for 2 min at 4°C. The cells were washed twice with ice-cold phosphate-buffered saline (PBS) and the pellets were stored at −20°C until assayed.
Cellular accumulation of Pt. Cell pellets from drug combinations were suspended in 0.5 ml 1% triton-X, held on ice while being sonicated. Total intracellular platinum contents were determined by graphite furnace AAS using a Varian SpectrAA-240 plus with GTA 120 atomic absorption spectrophotometer available in the Discipline of Biomedical Science The University of Sydney (24).
Platinum–DNA binding. DNA was isolated from cell pellet using H440050 JETQUICK Blood DNA Spin Kit/50 (Austral Scientific Pty Ltd.) according to the modified protocol of Bowtell (25). DNA content was determined by UV spectrophotometry (260 nm) (Varian 50 Bio UV-Visible spectrophotometer coupled with Varian 50 MPR Microplate Reader) and platinum content was determined by graphite furnace AAS. A260/A280 ratios were found to be between 1.75 and 1.8 for all samples ensuring its high purity (26) and the DNA concentration was calculated according to the equation: DNA concentration=Absorbance at 260 nm ×50 ng/μl.
DNA fragmentation. DNA isolated from A2780 and A2780cisR cells after their interaction with LH3 alone and in combination with genistein and resveratrol (as described earlier) using 0/0 h and 4/0 h sequences of administration in the case of genistein and 4/0 h sequence of administration in the case of resveratrol for 24 h, were subjected to agarose gel (2%) electrophoresis containing ethidium bromide as described previously. The amount of the DNA was kept constant (at 0.5 μg) for each drug. DNA bands were viewed under UV light and photographed as described previously.
Cellular glutathione. As a measure of the redox status of the cells, the levels of total glutathione (GSH and GSSG) and oxidized glutathione (GSSG) in A2780 and A2780cisR cells were determined for the 0/0 h and 4/0 h sequenced combinations of LH3 with genistein and curcumin. Drugs made in 10% RMPI-1640 serum-free medium were added to equal volumes of cell culture wells of a white wall clear-bottom 94-well plate containing exponentially growing A2780 and A280cisR cells (cell density=12×103 cells/well). Cells were left to incubate for 24 h. The media were aspirated out of the wells with minimal disturbance of the cell pellets and cells were washed with 200 μl of PBS, following which the levels of glutathione were determined using the GSH/GSSG-Glo™ Assay kit (Promega, Melbourne, Australia. The plate was read in a LUMIstar Omega luminometer (BMG LABTECH, Cary, USA).
Results and Discussion
Chemistry. Monofunctional LH3 was synthesized according to a modified Kauffman method (27). Cisplatin, used as a reference compound,was synthesized according to Dhara's method (28). Synthesis and characterization were fully described in the experimental section. Structure of LH3 is shown in Figure 1.
Molar conductivity. The limiting molar conductivity values (Λ0) for cisplatin and LH3 were found to be 136 and 316 Ω−1 cm2 mol−1, respectively. Higher molar conductivity for LH3 as compared to that for cisplatin is in line with the ionic nature of the compound. Whereas LH3 is an ionic compound, cisplatin is a polar molecule that produces a 1:2 electrolyte consisting of a di-positive cation and two chloride ions only upon hydrolysis. Thus, as cisplatin is administered intravenously, the molecule is expected to remain largely undissociated in blood serum, which has a high chloride concentration. This means cisplatin can cross the cell membrane by passive diffusion, although it is also known to be transported by carrier-mediated transport (29) and pinocytosis (30). In contrast, LH3 cations are likely to cross the cell membrane by carrier-mediated transport only (31).
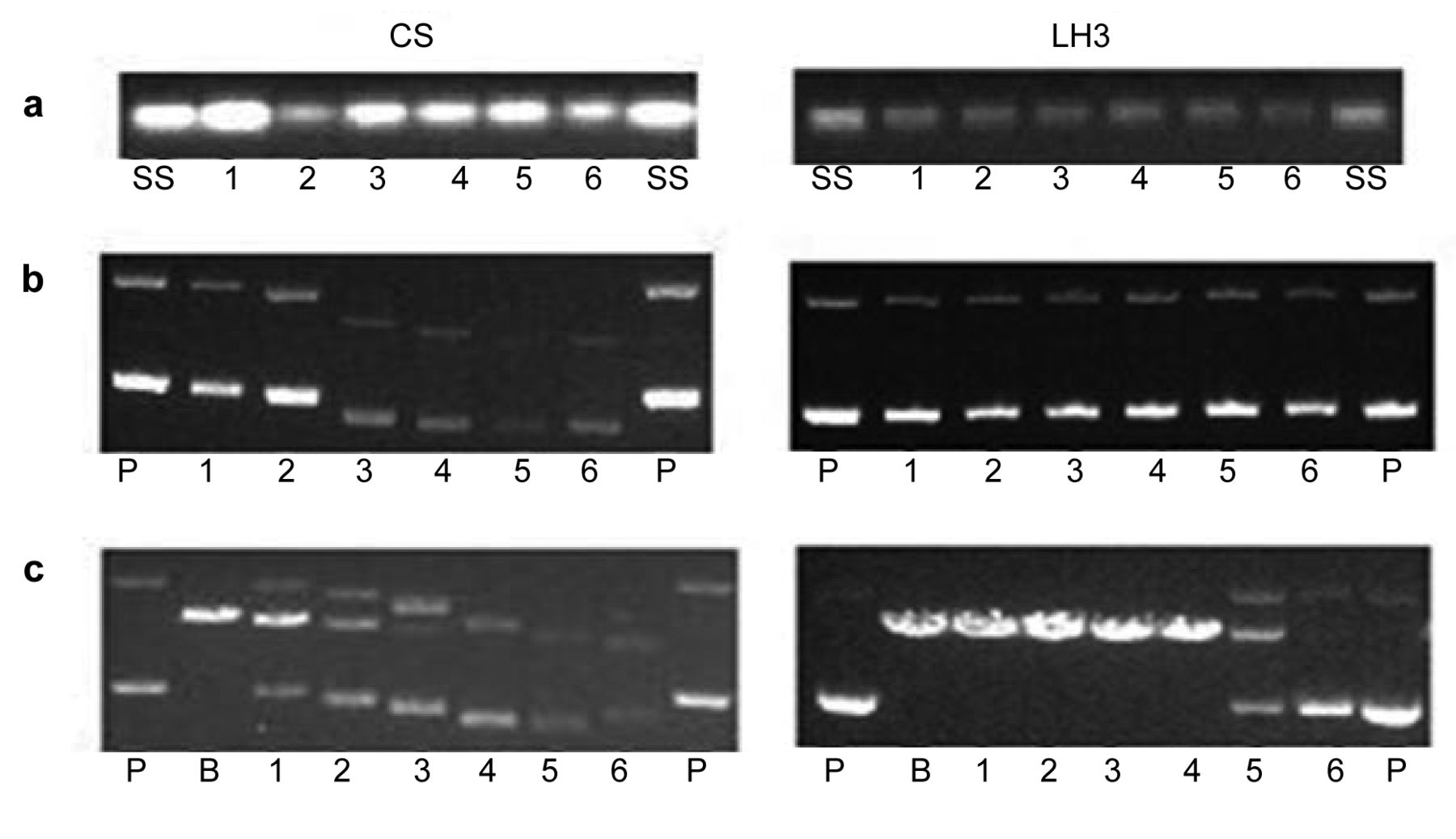
Electrophoretograms applying to the interaction of a: salmon sperm DNA; b: pBR322 plasmid DNA without BamH1 digestion; c: pBR322 plasmid DNA followed by BamH1 digestion with increasing concentrations of cisplatin (CS) and LH3. Lanes: SS: Untreated salmon sperm DNA; P: untreated pBR322 plasmid DNA; B: untreated and digested pBR322 DNA by BamH1 digestion; 1: 1.87 μM; 2: 3.75 μM; 3: 7.5 μM; 4: 15 μM; 5: 30 μM; 6: 60 μM.
Interaction with DNA. Figure 3 shows the electropho-retograms applying to the interaction of ssDNA, pBR322 plasmid DNA without BamH1 digestion and pBR322 plasmid DNA followed by BamH1 digestion, with increasing concentrations of cisplatin and LH3 ranging from 1.87 to 60 μM (lane 1 to 6) for 5 h at 37°C.
ssDNA. A single band was observed with both untreated and treated ssDNA. No noticeable change in intensity of the band due to interaction with LH3 indicates that the compound was not able to cause any observable damage to the ssDNA. The decrease in intensity of the ssDNA band with an increase in concentration of cisplatin indicates that cisplatin was able to cause damage to ssDNA.
pBR322 plasmid DNA. When pBR322 plasmid DNA was interacted with increasing concentrations of LH3 without BamH1 digestion, two bands corresponding to forms I and II were observed at all concentrations ranging from 0 to 60 μM. There was no noticeable change in either mobility or intensity of the bands. In contrast, when pBR322 plasmid DNA was interacted with increasing concentrations of cisplatin without BamH1 digestion, although two bands corresponding to forms I and II were observed, there was a marked change in both mobility and intensity of the bands with an increase in concentration. Decrease in separation between the bands and in intensity of the bands with an increase in concentration of cisplatin was indicative of conformational change in DNA and the occurrence of DNA damage. As noted earlier, cisplatin forms mainly intrastrand bifunctional 1,2–Pt(GG) and 1,2–Pt(AG) adducts with duplex DNA that cause significant bending of the DNA strand at and close to the binding site, thus bringing about changes in DNA conformation especially to the supercoiled form I of DNA (32, 33).
BamH1 digestion. BamH1 is a restriction enzyme that recognizes the sequence G/GATCC and hydrolyses the phosphodiester bond between adjacent guanine sites (34). pBR322 plasmid DNA contains a single restriction site for BamH1, which converts supercoiled form I and singly nicked circular form II pBR322 plasmid DNAs to linear form III DNA. When pBR322 plasmid DNA was interacted with increasing concentrations of LH3 followed by BamH1 digestion, only form III band was seen at low concentrations (1.87 μM to 15 μM) and at higher concentrations (30 μM and 60 μM), forms I, II and III bands were seen. The results indicate that LH3 was able to induce some change in DNA conformation. When pBR322 plasmid DNA was interacted with increasing concentrations of cisplatin followed by BamH1 digestion, forms I, II and III were observed at concentrations ranging from 1.87 μM to 7.5 μM and at higher concentrations (15 μM to 60 μM), forms I and II bands were observed. Mobility of the bands increased and intensity of the bands decreased (as did the separation between the bands) with an increase in concentration of the compound. Greater prevention of BamH1 digestion by cisplatin than LH3 indicates that cisplatin was more able to induce changes in DNA conformation than LH3, in line with the idea that bifunctional intrastrand 1,2–Pt(GG) and 1,2–Pt(AG) adducts formed by cisplatin can induce a greater change in DNA conformational than the monofunctional adducts formed by LH3. It is the protein recognition of DNA bending and unwinding that may be responsible for inducing cell death in downstream processes and may also be responsible for cell survival (e.g. through nucleotide excision repair of DNA lesions) resulting in drug resistance (35). As described later, damage to nuclear DNA in A2780 and A2780cisR cells was also investigated in this study.
The half-maximal Inhibitory concentrations (IC50) values and resistance factors (RF) of LH3, cisplatin (used as a reference compound), genistein, curcumin, resveratrol against the three cell lines: A2780, A2780cisR and A2780ZD0473R.
Growth-inhibitory effect of single drugs. Table I lists the IC50 values and the resistant factors (RF) of LH3, cisplatin (used as a reference compound), genistein, curcumin and resveratrol, as applied to the ovarian cancer cell lines: A2780 (cisplatin-sensitive; the parental cell line), A2780cisR (cisplatin-resistant) and A2780ZD0473R (picoplatin-resistant). The IC50 represents the drug concentration required for 50% cell kill and RF is defined as the ratio of the IC50 value in the resistant cell line to that in the parental cell line.
LH3 is about twelve-times more active than cisplatin against both the resistant cell lines, indicating that it is able to overcome platinum resistance. As LH3 contains three 8-hydroxyquinoline ligands compared to two ammonia ligands present in cisplatin, it can undergo distinctly different non-covalent interactions in terms of hydrogen bonding and stacking interactions with nucleobases in the DNA. Recently, it was reported that monofunctional cationic compounds containing bulky planaramine ligand can kill cancer cells with a greater efficacy than cisplatin (cisplatin) and oxaliplatin (31, 34).
Among the phytochemicals, curcumin was found to be most cytotoxic and resveratrol the least against all the three cell lines. Lower RF values for the phytochemicals (2 or less) and for LH3 (1 or less) than for cisplatin (about 12) indicate that these compounds were better able to induce cell death than cisplatin in the resistant cell lines.
Drugs in combination. The cell death from combinations of LH3 with genistein, curcumin, resveratrol and cisplatin in A2780, A2780cisR and A2780ZD0473R cancer cell lines was investigated as a function of the sequence of administration and the concentration. CIs as a quantitative measure of combined drug action, median-effect dose, shape (sigmoidicity), conformity (linear correlation coefficient r) are given in Table II. Figure 4 provides the CI values at the median-effect dose (ED50). It was found that for the combinations of LH3 with genistein and curcumin, the 0/0 h sequence of administration was most synergistic. In contrast, pre-treatment with genistein was required to result in synergistic outcome from the combinations of genistein with bi-functional platinums cisplatin and oxaliplatin (36). The results can be seen to indicate that the mechanisms underpinning synergistic outcome from combination of genistein with monofunctional platinum LH3 are different from those for its combination with cisplatin and oxaliplatin, even though both monofunctional and bi-functional platinums preferentially bind to N7 positions of guanine (34). However, uncertainty remains as to whether antitumour activity of LH3 and its analogues is limited to binding with DNA. Thus, any explanation for the strong synergism resulting from 0/0 h combination of LH3 with genistein, in terms of drug–DNA binding should be considered as being tentative. In support of this statement, it may be noted that the level of Pt–DNA binding resulting from 0/0 h combination of LH3 with genistein was not greater than that from LH3 alone. It is also important to stress that where platinum–DNA binding is a necessary first step towards cell kill, on its own it may not be sufficient to induce apoptosis as it is actually brought about by downstream processes in the cell cycle, where numerous proteins are involved. Whereas platinum resistance is associated with increased expression of transcription factor NF-κB and up-regulation of AKT and cyclooxygenase-2 (COX2) pathways thus promoting cell survival (37), genistein does the opposite (9, 38), including modulation of other signaling pathways such as protein-tyrosine kinase, metalloproteinases and BAX/B-cell lymphoma 2, (39, 40). Genistein has also an inhibitory effect towards protein-tyrosine kinase, which drives signal transduction pathways leading to tumor growth and progression to malignancy (41). Genistein being a polyphenol can scavenge free radicals (42), thus providing protection against oxidative damage to DNA and other biomolecules (42). This means that in presence of genistein, intracellular level of GSH would be higher, which in turn will result in increased deactivation of platinum before binding with DNA. Thus, it was found that platunum–DNA binding from antagonistic 4/0 h combination of LH3 with genistein was lower than that synergistic 0/0 h combination of LH3 with genistein in both parental A2780 and resistant A2780cisR cell lines.

Combination indices (CIs) applying to the 0/0 h, 0/4 h and 4/0 h combinations of LH3 with phytochemicals genistein (Gen), curcumin (Cur), resveratrol (Res) and cisplatin (CS) at the median effect dose (ED50) in A2780 (a), A2780cisR (b) and A2780ZD0473R (c) cell lines, calculated based on the pooled data from three to five individual experiments.

Agarose gel electrophoresis of DNA from A2780 cells (a) and (b) A2780cisR cells before and after interaction with LH3 and selected combinations with genistein (Gen)/resveratrol (Res). a: Lane 1: Control A2780, 2: LH3, 3: LH3+Gen 0/0 h, 4: LH3+Gen 4/0 h, 5: Control A2780; b: Lane 1: Control A2780cisR, 2: LH3, 3: LH3+Gen 0/0 h, 4: LH3+Gen 4/0 h, 5: LH3+Res 4/0 h, 6: Control A2780cisR.
Dose–effect parameters applying to combinations of LH3 with genistein, curcumin, resveratrol and cisplatin in the A2780, A2780cisR and A2780ZD0473R cell lines.
Platinum accumulations in A2780 and A2780cisR cells resulting from treatment with combinations of LH3 with Gen, Cur, Res and Cis (at 5 μM for LH3 and 25 μM for Gen, Cur, Res and Cis) for 24 h at 37°C, given using 0/0 h, 0/4 h and 4/0 h sequences of administration.
Level of platinum–DNA binding (nmol Pt per mg of DNA) in A2780 and A2780cisR cells resulting from their interaction with combinations of LH3 with Gen, Cur, Res and Cis (at 5 μM for LH3 and 25 μM for Gen, Cur, Res and (is) for 24 h at 37°C, given using 0/0 h, 0/4 h and 4/0 h sequences of administration.
In the combinations of LH3 with curcumin, the bolus administration produced synergistic outcomes in all three cell lines (more so in the resistant cell lines), whereas 0/4 h and 4/0 h sequences of administrations were antagonistic to additive. The results are different from those resulting from the combination of curcumin with cisplatin and oxaliplatin in the same ovarian cancer cells, where prior incubation of cells with curcumin for a short period served to stimulate them towards the cell killing effect due to bi-functional platinums (43). The enhanced cell kill due to the presence of curcumin cannot be attributed to the antioxidant role played by the phytochemical. Because when it so acted, it would serve to reduce the oxidative stress resulting in sparing of cellular antioxidants such as glutathione. Increased level of platinum–DNA binding observed with antagonistic 4/0 h combination of curcumin and LH3 in A2780cisR cell line indicates that the above explanation could not be true. As stated earlier, platinum resistance and enhancement of platinum action due to combination with phytochemicals appear to be related to NF-κB. Whereas resistance to platinum drugs is associated with aberrant activation of NF-κB, the presence of curcumin could serve to dampen its expression. It has been reported that curcumin potentiates the antitumour effects of gemcitabine and fluorouracil (5-FU) by inhibiting gemcitabine-induced NF-κB activation (and its downstream targets), leading to inhibition of cell proliferation, angiogenesis, and invasion in an orthotopic model of pancreatic cancer (44). It was also found that curcumin enhanced the antitumour activity of capecitabine through modulation of cyclin D1, COX2, matrix metallopeptidase-9 and C-X-C chemokine receptor type 4 (45) that of oxaliplatin in colon cancer cells through down-regulation of COX2 (46) and modulation of EGFR and insulin-like growth factor 1 receptor (47).
Levels of reduced (GSH) and oxidized (GSSG) glutathione in relative luminescence unit (RLU) and the GSH/GSSG ratio before and after treatments with: LH3 alone and 0/0 h and 4/0 h combinations of LH3 with Gen and Cur in the ovarian A2780 and A2780cisR cancer cell lines.
Besides NF-κB, other mediators are also likely to be involved in chemosensitization due to curcumin, especially as applied to cisplatin chemotherapy. For example, the pathway which is responsive to DNA damage has been given as a reason for cisplatin resistance in ovarian tumors (13). Curcumin has been shown to inhibit this pathway and sensitize CAOV3 and SKOV3 ovarian cancer cells to cisplatin-induced apoptosis (14, 15).
With resveratrol, all combinations with LH3 were found to produce antagonistic to additive effects, irrespective of the sequence of administration in all the three ovarian cell lines. The results indicate that resveratrol may not be a suitable candidate for combination with monofunctional platinums. In line with the results of the present study, resveratrol was reported to produce antagonistic effects when applied in combination with paclitaxel, vincristine and daunorubicin to a number of tumour models (17, 18). This was explained as being due to induction of cell-cycle arrest at the S phase, thus preventing cells from entering the G2/M phase where paclitaxel exerts its effect (19). Supporting observations were also made in studies on bladder (48) and breast cancer (49) cells. However, none of the studies provided an explanation as to how the cells proliferated after S-phase arrest. It is probable that downstream processes responsible for cell death have interfered with each other to produce antagonistic outcomes.
The key regulators of resveratrol-mediated chemosensitization appear to be the anti-apoptotic and cell survival gene products (50) such as BCL2 (51) NF-κB, signal transducer and activator transcription factor 3 (5). It is also linked to the regulation of COX expression (52). Recently it has been reported that NF-κB may play a paradoxical role in ovarian cancer. It can function as a biphasic regulator suppressing as well as enhancing ovarian cancer growth through regulation of MAPK and induction of apoptosis (53). Whereas NF-κB activation is required for cisplatin-induced apoptosis in head and neck squamous carcinoma cells (54, 55), activation of NF-κB signaling by IKKβ increases aggressiveness of ovarian cancer (56) so that inhibition of NF-κB activity would enhance apoptosis and inhibit cell growth in type-2 chemotherapy-resistant epithelial ovarian cancer cells (53). The conflicting role of NF-κB in apoptosis indicates that the effects of many cellular signaling molecules are dependent upon cell content and/or stress condition (57). Thus, it is possible that combinations of LH3 and resveratrol favour the stimulation of NF-κB leading to reduced cell death. Lastly, both the dose–response curves (Table II) and CI values (Figure 4) showed that irrespective of sequence of administration, combinations of LH3 with cisplatin produced antagonistic to additive outcomes in cell kill in all the three cell lines. Similarly, it was reported that when monofunctional platinum pyriplatin was combined with cisplatin, additive to antagonistic effects were produced in HT29 and OVCAR-3 cell lines (31). Like cisplatin and oxaliplatin, pyriplatin induces cell-cycle arrest at G2-M phase in ovarian HCT-116 and MCF7 cell lines with wild-type p53 status (31). Thus, the observed antagonism from the combination of LH3 with cisplatin is not unexpected when we note that it may be causing cell-cycle arrest at the same phase.
Cellular platinum accumulation. Table III provides platinum accumulation in A2780 and A2780cisR cells after their interaction with combinations of LH3 with genistein, curcumin, resveratrol and cisplatin (at 5 μM for LH3 and 25 μM for genistein, curcumin, resveratrol and cisplatin) for 24 h at 37°C, administered using 0/0 h, 0/4 h and 4/0 h sequences. Synergistic 0/0 h combinations of LH3 with genistein and curcumin were found to be associated with high platinum accumulation in A2780cisR cell line. However, antagonistic 4/0 h combination of LH3 with curcumin was associated with even higher Pt accumulation in the A2780cisR cell line. The results can be seen to indicate that combined drug action associated with the combinations of LH3 with genistein and curcumin and the levels of intracellular Pt accumulation do not follow any clear trend except that a greater synergism often resulted from concurrent administration or preteatment with the phytochemicals. Characterization of key proteins responsible for platinum resistance in ovarian tumor models and their modulation due to treatment with drug combinations may provide a clearer picture. Even then the presence of crosstalk between pro-apoptotic and anti-apoptotic pathways may complicate the situation making difficult interpretation of results. The results from the combinations of LH3 with resveratrol appear to be even more complex. For example, both additive 0/0 h and slightly antagonistic 4/0 h combinations of LH3 with resveratrol were found to be associated with high intracellular Pt accumulation in the A2780cisR cell line. In spite of the observed complexity, it can be seen that for almost all combinations of LH3 with the phytochemicals and cisplatin, platinum accumulation in the resistant A2780cisR cell line was greater than in the parental A2780 cell line, indicating that the presence of the phytochemical had served to enhance uptake of platinum in the resistant cell line.
Platinum–DNA binding level. Table IV provides the level of platinum–DNA binding in A2780 and A2780cisR cells after their interaction with combinations of LH3 with genistein, curcumin, resveratrol and cisplatin (at 5 μM for LH3 and 25 μM for genistein, curcumin, resveratrol and cisplatin) for 24 h at 37°C, administered using 0/0 h, 0/4 h and 4/0 h sequences.
The results show that for the combinations of LH3 with cisplatin administered to A2780 and A2780cisR cell lines, in the parental cell line, higher level of Pt–DNA binding resulted from antagonistic 0/0 h sequence of administration and in the resistant cell line, resulting from antagonistic 4/0 h sequence of administration. The results can be seen to provide support to the idea that the cell killing effect due to LH3, whether alone or in combination, could not be the result of simply drug–DNA binding.
As discussed earlier, even for cisplatin and its analogues where binding with DNA is considered to be an essential step in initiating the course of events towards apoptosis, cell death is actually brought about by downstream processes in the cell cycle where numerous proteins are involved. As discussed by Sadler (35), protein recognition is the key requirement for both the cell death and the escape from it. As applied to the combinations of LH3 with genistein, synergistic 0/0 h sequence of administration resulted in slightly higher Pt–DNA levels than antagonistic 4/0 h sequence of administration in both A2780 and A2780cisR cell lines in line with the trend observed for intracellular platinum accumulation. As applied to the combinations of LH3 with curcumin, both 0/0 h and 4/0 h sequences of administration were found to be associated with high Pt–DNA binding levels in both A2780 and A2780cisR cell lines. Although the high Pt–DNA binding level from synergistic 4/0 h combination of LH3 and curcumin (especially in the resistant A2780cisR cell line) can be seen to be in line with enhanced cell kill, antagonistic 4/0 h combination of LH3 with resveratrol was also associated with high Pt–DNA binding level in the resistant A2780cisR cell line. As discussed earlier, the results from combinations of LH3 with resveratrol appear to be more complex, defying any clear trend.
DNA fragmentation. Figure 5 shows the bands from agarose gel electrophoresis of DNA isolated from A2780 and A2780cisR cells before and after their interaction with LH3 and its selected combinations with genistein and resveratrol. The observed elongation of DNA band indicates that LH3 alone and in combination with genistein and resveratrol was able to cause damage to DNA present in A2780 and A2780cisR cells. Most pronounced streaking of the DNA band in the case of strongly synergistic 0/0 h combination of LH3 with genistein in A2780cisR cell line can be seen to indicate that the DNA damage was in line with the high degree of synergism.
The loss in intensity of the DNA band observed in lanes 4 and 5 of Figure 5 (applying to the interaction of A2780cisR cells with antagonistic 4/0 h combinations of LH3 with genistein and resveratrol), may be indicative of a decrease in DNA fluorescence rather than damage to the DNA as pre-treatment with genistein and resveratrol (both of which can act as intercalators) might have served to reduce intercalation of ethidium bromode (58). More importantly the results may also point-out the limitations of correlating DNA damage in surviving cells with cell death.
Cellular glutathione levels. In the treatment of cancer cells with tumor-active phytochemicals (whether alone or in combination with a targeted chemotherapeutic), it is expected to exist a dynamic interplay between dual functions of the phytochemicals namely toxicity and protection of cells against oxidative stress. In this study, cellular GSH and GSSG levels were determined before and after treatment of ovarian A2780 and A2780cisR cancer cells with LH3-alone and in combination with genistein and curcumin. Table V provides the levels of both reduced and oxidized glutathione (GSH and GSSG) in relative luminescence unit (RLU) and the GSH/GSSG ratio before and after treatment of A2780 and A2780cisR cells with 0/0 h and 4/0 h combinations of LH3 with genistein and curcumin.
The results show that the level of GSH was greater in the resistant A2780cisR cell line than in the parental A2780 cells both before and after treatment with LH3 and its combinations with selected phytochemicals, thus giving support to the idea that elevated expression of GSH can serve as a mechanism of platinum resistance in tumour models (59, 60). The level of GSSG was also higher in the resistant cells, so that the ratio GSH:GSSG was actually lower for the resistant A2780cisR cell line than for the parental A2780 cell line.
When cells were treated with 0/0 h and 4/0 h combinations of LH3 and curcumin, the resulting levels of both the reduced and oxidised glutathione were found to be greater in the parental A2780 cell line as compared to the values found before any drug treatment and also after treatment with LH3 alone. The higher levels of GSH found in A2780 cells after treatment with 0/0 h and 4/0 h combinations of LH3 and curcumin can be explained in terms of antioxidant role played by curcumin so that GSH was spared. Higher levels of GSH and GSSG were found in untreated A2780cisR cells than in untreated A2780 cells (about 2.9-times as large for A2780 cells and 3.6-times as large for A2780cisR) so that the effect of drug treatment on levels of GSH and GSSG was less significant for the resistant cells. When A2780 and A2780cisR cells were treated with LH3 alone, unexpectedly, the resulting levels of GSH and GSSG were slightly greater than the corresponding values found in untreated cells. The results may indicate that monofunctional platinum LH3 did not undergo any significant binding to GSH. Alternatively in the presence of LH3, extrusion of both forms of glutathione through efflux transporters such as the multidrug resistance-associated proteins may have decreased (61).
For treatment of cells with 0/0 h and 4/0 h combinations of LH3 and genistein, it was found that the levels of GSH and GSSG were slightly lower in the parent A2780 and resistant A2780cisR cells than the values found in untreated cells. Slightly lower levels of GSH found after treatment of A2780 and A2780cisR cells with the combinations of genistein and LH3 in contrast to the higher levels found after treatment with combinations of LH3 and curcumin can be seen to indicate that genistein may be less potent than curcumin as an antioxidant.
In actual fact, GSH levels in A2780 cells resulting from treatment with combinations of LH3 and genistein were lower than the value found after treatment with LH3-alone, whereas the level of GSSG was higher. Although a higher level of GSSG can be due to decreased extrusion of GSSG through efflux transporters, the above results indicate that in presence of LH3, genistein acted more likely as a pro-oxidant (61). Almost additive 4/0 h combination of LH3 with curcumin in the A2780 cell line was found to be associated with the largest GSH:GSSG ratio, whereas synergistic 0/0 h combination of LH3 with curcumin in the A2780cisR cell line was associated with the lowest value. The results can be seen to indicate that for the combinations of LH3 with curcumin administered to A2780 and A2780cisR cells, a decrease in GSH:GSSG ratio led to increased cell kill, cell death had occurred via extrinsic pathway mediated by oxidative stress.
Conclusion
Distinctly different structural features of LH3 compared to cisplatin, and its pronounced cytotoxicity (especially against the resistant cell lines) clearly indicate that the compound was better able to overcome mechanisms of resistance operating in the ovarian A2780cisR and A2780ZD0473R cancer cell lines. Binary combinations of LH3 with genistein, curcumin and resveratrol were found to produce sequence- and concentration-dependant synergism in the A2780, A2780cisR and A2780ZD0473R ovarian cancer lines, with the bolus combinations generally being the most synergistic.
Future Directions
LH3 has the potential for development as a novel platinum-based anticancer drug provided it shows activity in vivo. Thus, in vivo studies using suitable animal studies are needed. Proteomic studies and studies on DNA microarray may provide further information on the mechanism of action of LH3 and its combinations with phytochemicals.
Acknowledgements
The Authors are thankful to Dr Ian Luck of the School of Chemistry, The University of Sydney, Australia for recording 1H NMR spectra. Laila Arzuman is grateful to The University of Sydney for the award of a UPA Scholarship. This project is supported by Biomedical Science Research Initiative Grant and Biomedical Science Cancer Research Donation Fund.
- Received July 22, 2014.
- Revision received September 4, 2014.
- Accepted September 9, 2014.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Protein Expression Patterns in ovarian cancer cells Associated with Monofunctional Platinums Treatment
- Synthesis of tris(quinoline)monochloroplatinum(II) Chloride and its Activity Alone and in Combination with Capsaicin and Curcumin in Human Ovarian Cancer Cell Lines
- Monofunctional Platinum-containing Pyridine-based Ligand Acts Synergistically in Combination with the Phytochemicals Curcumin and Quercetin in Human Ovarian Tumour Models