


Abstract
The prognosis for patients with glioblastoma is very poor, despite intensive treatment, including surgery and chemoradiotherapy. Wilms' tumor 1 (WT1) is expressed in most glioblastoma samples, and immunotherapy targeting WT1 has proven to be effective in recurrent glioblastoma. However, the functional roles of WT1 in glioblastoma are not clear. To examine the functional roles of WT1 in glioblastoma, glioblastoma cell lines with reduced WT1 expression were generated using short hairpin RNA(shRNA)-expressing lentivirus. Proliferation of WT1-knockdown glioblastoma cells was significantly slower than control cells with high WT1 expression. In addition, apoptosis was increased in WT1-knockdown glioblastoma cells. Furthermore, WT1-knockdown glioblastoma cells, and control glioblastoma cells were intra-cranially injected into immunodeficient mice. In vivo tumor growth of WT1-knockdown glioblastoma cells was significantly reduced compared to control glioblastoma cells. These results show that WT1 is involved in glioblastoma cell proliferation and apoptosis and that this protein has oncogenic roles in glioblastoma.
Glioblastoma is one of the most frequent malignancies in the central nervous system. Despite intensive treatment including surgery, radiation, and chemotherapy, the prognosis is still very poor, and the median survival time is only 12-15 months (1). To improve the prognosis of patients with glioblastoma, various therapies have been tested. Wilms' tumor 1 (WT1) is a therapeutic target in glioblastoma cells, and WT1 peptide vaccine immunotherapy targeting WT1 has proven effective in recurrent glioblastoma (2).
WT1 is expressed in various types of hematological malignancies and solid tumors (3-11). The WT1 gene was first defined as a tumor suppressor gene (12-17). However, accumulating evidence suggests that the WT1 gene has an oncogenic function in tumorigenesis. The growth of WT1-expressing cancer cells is inhibited by WT1 antisense oligomer (18, 19) and WT1-specific shRNA (20). Furthermore, overexpression of WT1 promotes cell growth (21-23), migration, and invasion (24). Overexpression of WT1 also inhibits apoptosis (25) and induces tumorigenicity in leukemia (26). However, functional roles of WT1 in glioblastoma have not been extensively studied.
In the present study, we investigated the functional roles of WT1 in glioblastoma. We first examined whether WT1 is involved in proliferation and apoptosis in glioblastoma cells. In addition, we also investigated whether WT1 is involved in tumorigenicity in an intra-cranial xenograft model.
Materials and Methods
Establishment of WT1 shRNA glioblastoma cell lines. We obtained the glioblastoma cell lines U87MG and U251 from the American Type Culture Collection (ATCC, Manassas, VA, USA). To establish stable WT1-knockdown cells, Mission shRNA (Sigma Aldrich, St. Louis, MO, USA) carrying shRNA sequence against WT1 (5’-CCGGGAAACCATACCAGTGTGACTTCTCGAGAAGTCACACTG GTATGGTTTCTTTTT-3’) was used. Lentivirus was produced by trasfection of the lentiviral vector with the gag-pol-expressing vector and the VSV-G envelope-expressing vector. Viruses were concentrated by centrifugation with PEG-it (System Bioscience, Mountain View, CA, USA). U87MG and U251 glioblastoma cells were infected with lentivirus carrying WT1-shRNA. Knockdown of WT1 was confirmed with Real-Time Polymerase Chain Reaction (PCR).
Quantitative PCR. Total RNA was extracted from the glioblastoma cell lines U87MG and U251 using Trizol (Invitrogen Life Technologies Inc., Carlsbad, CA, USA) according to the manufacturer's instructions. cDNA was generated using the Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI, USA) and then subjected to quantitative PCR with SYBR green in an ABI-7900 HT instrument (Applied Biosystems, Life Technologies Inc.). To measure Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and WT1 expression, the following primers were used: for GAPDH: 5’-GCCAAAAGGGTCATCATCTC-3’ (sense), 5’-GTAGAGGCAGGGATGATGTTC-3’ (antisense); for WT1: 5’-GATAACCACACAACGCCCATC-3’ (sense), 5’-CACACGTCGCACATCCTGAAT-3’ (antisense).
Gene expression array analysis. RNA was isolated from control and WT1-knockdown glioblastoma cell lines (U87MG and U251) using Trizol. cDNA was synthesized by reverse transcriptase reaction. cDNA from each type of cells was analyzed with a Taqman gene expression array kit (Applied Biosystems, Life Technologies Corporation).
Cell growth assay. Cells (3×104 cells/well; U87MG and U251 control cells, U87MG and U251 WT1 knockdown) were plated in a 12-well plate (Costar, Corning Life Sciences, Corning, NY, USA). These cell lines were cultured in DMEM+10%FBS media. The number of cells was counted using a cytometer following Trypan blue staining at days 0, 2, and 4. These experiments were repeated three times.
Apoptosis assay. We used an Annexin-V-Fluos Staining kit (Roche Applied Science, Indianapolis, IN, USA) following the manufacturer's instructions. Cells (5×104) were plated in a 4-well culture slide (Becton Dickinson Bioscience, San Jose, CA, USA) and incubated in 5% CO2 for 24 h After incubation, we counted the number of apoptotic cells using a fluorescence microscope (Keyence, Osaka, Japan). We randomly selected four magnified fields and calculated the average number of apoptotic cells per field. These experiments were repeated three times.
Intracranial xenograft model. Newborn Rag2−/−γc−/− mice (kindly donated by Irving Weissman, MD, Stanford University) were used as recipients. Pups were anesthetized on ice, and glioblastoma cells (2×105) in 2 μl Phosphate Buffered Saline (PBS) were injected into the right lateral ventricle with a stereotactic injector (Stoelting, Wood Dale, IL, USA). The tumor injection point was as follows; 2 mm forward of bregma, 1 mm right, 2 mm deep. We sacrificed mice when they showed neurological symptoms or otherwise at day 60.
Immunohistochemical analysis. Formalin-fixed paraffin-embedded (FFPE) tumor samples were obtained from mice with U87MG glioblastoma cells transduced with shRNA against WT1 and cells transduced with control shRNA. FFPE samples were cut at 6-μm thickness with a microtome. Sections mounted on slides were heated in a microwave oven for antigen retrieval. We used mouse anti-human monoclonal antibody against WT1 (at a dilution of 1:50) and Ki67 (at a dilution of 1:50) (Dako, Glostrup, Denmark) as primary antibodies. The linked Streptavidin-Biotin detection method was selected, and Histofine simple stain MAX-PO (Multi) (Nichirei Bioscience, Tokyo, Japan) was used as a secondary antibody. Specimens were reacted and visualized with diaminobenzidine (Dojindo, Kumamoto, Japan).
Statistical analysis. Student's t-test was used to determine statistical significance for the in vitro experiments. For the analysis of in vivo experiments, Kaplan-Meier analysis was used. For all analyses, differences were defined as statistically significant when the p-value was less than 0.05.
Results
WT1 plays a role in cell proliferation in glioblastoma cells in vitro. We first examined whether WT1 was involved in cell proliferation in vitro. Cell proliferation was compared in U87MG and U251 glioblastoma cells transfected with shRNA against WT1 and those transfected with control shRNA (Figure 1A). The total numbers of cells were 39±7.8×104, 19±3.5×104, 22.5±3.5×104, and 3.4±0.5×104 cells in control shRNA-transfected U87MG cells, anti-WT1 shRNA-transfected U87MG cells, control shRNA-transfected U251 cells, and anti-WT1 shRNA-transfected U251cells, respectively (p<0.05) at day 4. We found that cell proliferation was significantly reduced in U87MG and U251 cells transfected with shRNA against WT1 (Figure 1B). These results showed that WT1 is involved in proliferation of glioblastoma cells.
WT1 promotes glioblastoma formation in a xenograft model. We next examined the effect of WT1 on glioblastoma progression in vivo. WT1-knockdown U87MG cells transduced with shRNA against WT1 or control U87MG cells transduced with control shRNA were intra-cranially injected into the right ventricle of newborn Rag2−/−γc−/− pups. The difference in the survival curve was significant (p<0.05) between xenograft mice injected with WT1-knockdown U87MG cells and those injected with control U87MG cells (Figure 2A). All xenografted mice (n=6 in total) injected with control U87MG cells died of glioblastoma development within 40 days after tumor injection, whereas none of the xenografted mice injected with WT1-knockdown U87MG cells had died of glioblastoma development by post-transplant day 40 (Figure 2B), thus demonstrating that WT1-knockdown significantly inhibits glioblastoma growth in this model.
All xenografted mice with WT1-knockdown U87MG cells were sacrificed at day 60. Only one mouse had developed a glioblastoma. We then examined the difference between resected tumors from mice with control U87MG cells and those from mice with WT1-knockdown U87MG cells. The number of tumor cells per 400× field was 175.2±10.5 cells in tumors from mice with control U87MG cells and 99.8±12.7 cells in tumors from mice with WT1-knockdown U87MG cells. Thus, the cellular density in tumors from mice with control U87MG cells was significantly greater than that in mice with WT1-knockdown U87MG cells (Figure 2C). Immunohistochemical analysis showed that the Ki-67 labeling index was 30.5% in tumors from mice with control U87MG cells and 14.8% in tumors from mice with WT1-knockdown U87MG cells (Figure 2D). These data indicate that WT1 is involved in cell proliferation and tumor formation in vivo.
WT1 is involved in glioblastoma progression by regulating apoptosis. We next investigated differences in gene expression between U87MG and U251 cells transduced with shRNA against WT1 and cells transduced with control shRNA. We isolated RNA from these four cell types and performed real-time PCR to examine up-regulated or down-regulated genes in glioblastoma cells transduced with shRNA against WT1. We found that apoptosis-related genes such as Mitogen-activated protein kinase kinase kinase 5 (MAP3K5), phosphoinositide-3-kinase, catalytic, subunit alpha (PIK3CA), and p53 were up-regulated in both U87MG and U251 cells transduced with anti-WT1 shRNA, compared with those transduced with control shRNA (Table I).

Wilms' tumor-1 (WT1) is involved in cell proliferation in glioblastoma cells. A: Quantitative Real-Time Polymerase Chain Reaction from U87MG cells transduced with control shRNA, U87MG cells transduced with WT1 shRNA, U251 cells transduced with control shRNA, and U251 cells transduced with WT1 shRNA. B: Cell proliferation rate of U87MG cells transduced with control shRNA and U87MG cells transduced with WT1 shRNA. C: Cell proliferation rate of U251 cells transduced with control shRNA and U251 cells transduced with WT1 shRNA.
List of genes up-regulated or down-regulated in U87MG or U251 cells transduced with WT1 shRNA cells compared to the same cells transduced with control shRNA.
We next used an Annexin-V-Fluos Staining kit to investigate whether WT1 is involved in apoptosis of glioblastoma cells. The numbers of apoptotic cells per 30× microscopic field were 2.5±0.6, 15.3±5.7, 3.0±1.7, and 9.0±1.0 cells in U87MG cells with control shRNA, U87MG cells with anti-WT1 shRNA, U251 cells with control shRNA, and U251 cells with anti-WT1 shRNA, respectively (Figure3A and B) (p<0.05). These results show that apoptosis was promoted in both U87MG and U251 cells transduced with shRNA against WT1 compared with those transduced with control shRNA, suggesting that WT1 is involved in glioblastoma tumorigenicity by regulating apoptosis.
Discussion
In the present study, we showed that WT1 is involved in cell proliferation and apoptosis in glioblastoma. Furthermore, silencing of WT1 inhibits xenograft tumor formation in an immune-deficient mouse model. Gene expression analysis and p53 protein expression analysis suggest that WT1 may regulate glioblastoma progression by regulating expression of the tumor-suppressor p53.
Functional roles of WT1 in glioblastoma have been reported in several studies (7, 20, 27, 28). Oji et al. reported that the growth of U87MG is inhibited by a WT1 antisense oligomer (7), suggesting that WT1 is involved in cell proliferation in glioblastoma. Our results are consistent with theirs. Tatsumi et al. reported that WT1 is involved in apoptosis in the A172 glioblastoma cell line (20). They also examined apoptosis using the annexin V assay. Our results confirm that WT1 is involved in glioblastoma cell apoptosis. Clark et al. reported that WT1 is involved in cell proliferation in U251 cells, and U251 cells transduced with shRNA against WT1 had significantly lower tumorigenicity in a subcutaneous nude mouse model (28).

Wilms' tumor-1 (WT1) is involved in glioblastoma tumorigenicity in vivo. A: Newborn Rag2−/−γc−/− pups were injected with U87MG cells transduced with control shRNA, or U87MG cells transduced with WT1 shRNA. A Kaplan-Meier survival curve for each group is shown. B: Representative images of tumors derived from U87MG cells transduced with control shRNA or with WT1 shRNA. C: Hematoxylin and eosin staining and immunohistochemical staining of tumor samples from mice injected with U87MG cells transduced with WT1 shRNA or with control shRNA. D: Immunohistochemical staining for Ki-67 in tumor samples from mice injected with U87MG cells transduced with WT1 shRNA or with control shRNA.
However, several controversies exist in the functional roles of WT1 in glioblastoma. Chidambaram et al. reported that silencing of WT1 reduced invasiveness of U1242 cells, but had no effect on cell proliferation in vitro (29). Clark et al. also reported that apoptosis is not different between T98G cells transduced with shRNA against WT1 compared with those transduced with control shRNA in vitro (28). Their results are different from these of our study. The difference in functional roles of WT1 in cell proliferation and apoptosis may be due to the different glioblastoma cell lines used for the experiments. These results suggest that the functions of WT1 on cell proliferation and apoptosis in vitro might be different in various glioblastoma cell lines.
In our study, we first showed that WT1 is involved in glioblastoma tumorigenicity in an intracranial xenograft model. Clark et al. also reported that WT1 is involved in glioblastoma tumorigenicity in vivo (28). However, they used a subcutaneous xenograft model, and thus, our intracranial in vivo assay better-reflects the functional roles of WT1 in glioblastoma in vivo. Furthermore, we also first analyzed tumor samples from mice with U87MG cells transduced with shRNA against WT1 and found that the Ki-67 labeling index was significantly reduced compared to tumors of control-transduced U87MG cells. These findings also support a functional role for WT1 in cell proliferation. Clark et al. also reported that the Ki-67 labeling index from subcutaneous tumors with U251 cells transduced with shRNA against WT1 was not different from that in tumors with U251 cells transduced with control shRNA (28). In their study, WT1 expression in tumors from U251 cells transduced with shRNA against WT1 was not different from U251 cells transduced with control shRNA, and thus, the quality of WT1-knockdown was not sufficient to conclude a difference in the Ki-67 index.
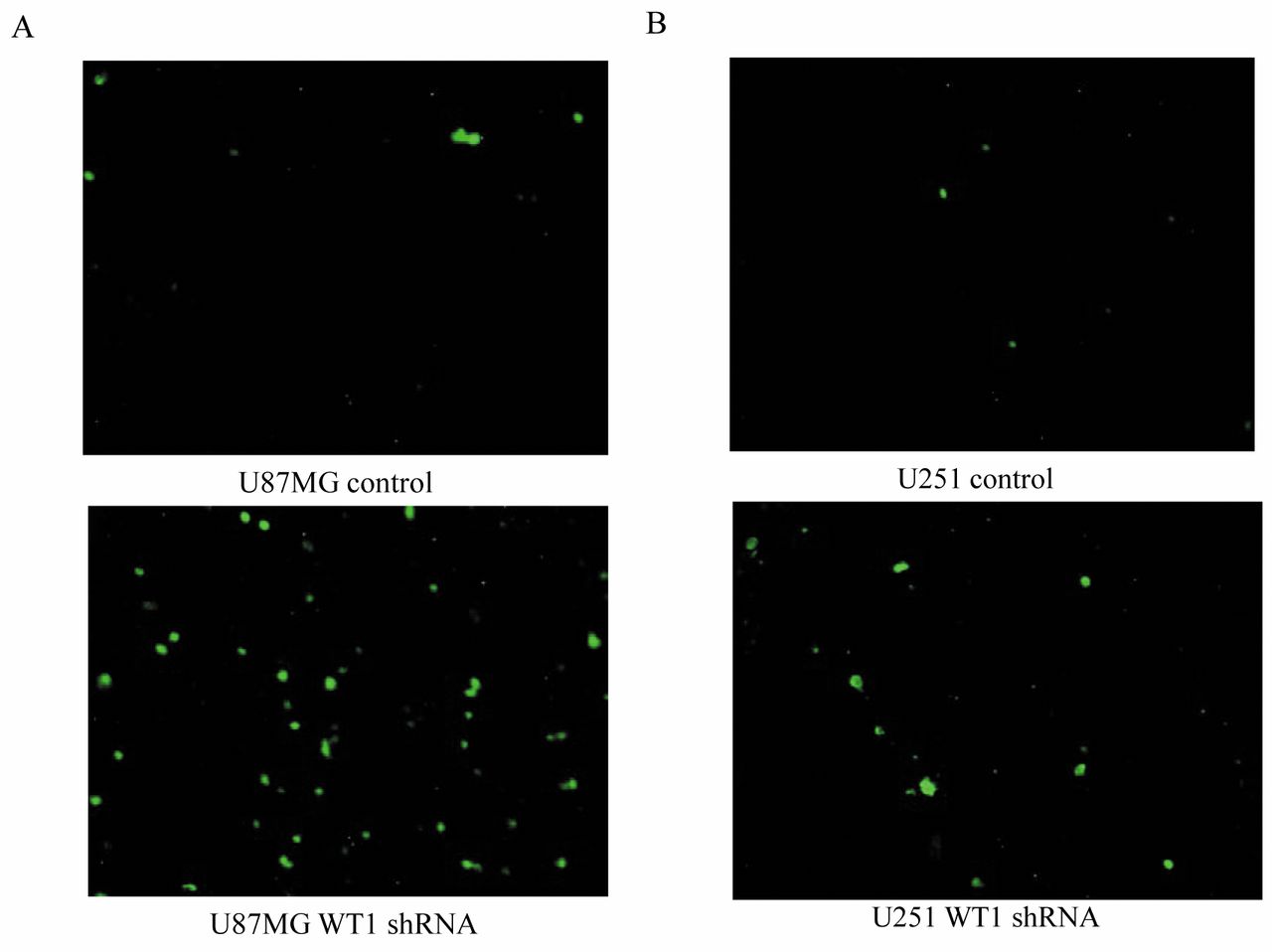
Wilms' tumor-1 (WT1) is involved in apoptosis of glioblastoma cells. A: Representative images of annexin V-positive U87MG cells transduced with control shRNA or with WT1 shRNA. B: Representative images of annexin V-positive U251 cells transduced with control shRNA or with WT1 shRNA.
In the present study, we used immune-deficient mice, and thus, silencing of WT1 reflected the functional roles of WT1 in glioblastoma cells. However, the antitumor immunity in an immune-competent environment may be non-functional when WT1 expression in glioblastoma cells is low. In fact, Chiba et al. reported that patients with glioblastoma with intermediate WT1 expression had the most favorable prognosis among patients receiving WT1 peptide vaccination (30). Thus, when we consider WT1 as a therapeutic target in patients with glioblastoma, complete silencing of WT1 in glioblastoma may lead to poor antitumor immunity, leading to less favorable prognosis.
Collectively our study suggests that WT1 functions as an oncogene in glioblastoma, in part by regulating cell proliferation and apoptosis.
In conclusion, WT1 is involved in glioblastoma cell proliferation and apoptosis, plays oncogenic roles in glioblastoma, and thus, may be a feasible molecular target for the treatment of glioblastoma.
- Received November 13, 2013.
- Revision received December 2, 2013.
- Accepted December 3, 2013.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.