


Abstract
Aim: To evaluate the in vitro cytotoxicity of oxoglaucine (OG) complexes: [Sm(OG)2(NO3)3]•H2O (1), [Eu(OG)2(NO3)3]•1.5CH3OH (2) and [Er(OG)2(NO3)3]•H2O (3) through comparison to oxoglaucine and lanthanide salts. Materials and Methods: The reactions of OG with corresponding lanthanide salts gave rise to complexes 1-3. The crystal structures of complexes 1-3 were determined by single-crystal X-ray diffraction analysis. The in vitro cytotoxicity of oxoglaucine and complexes 1-3 against five human cancer cell lines were evaluated by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium Bromide (MTT) method. Results: Complexes 1-3 have similar mononuclear structures. The 50% inhibitory concentration (IC50) of complex 1 against SGC7901 cells was 32.1 μM; that of complex 2 against MCF-7 cells was 3.2 μM; those of complex 3 on HeLa and MCF-7 cells were 8.3 and 1.4 μM, respectively. Conclusion: The three OG-lanthanide complexes exhibited significantly enhanced cytotoxicity vs. OG and corresponding lanthanide salts.
Since the success of cisplatin and related platinum complexes as anticancer agents, the development of other active transition metal anticancer complexes with better efficiency has attracted increasing interest of many bioinorganic chemists (1-4). Of special attention are lanthanide complexes, which manifest antitumour activity with the potential of becoming future anticancer drugs (5, 6). In the past two decades, a number of lanthanide complexes have been synthesized and their cytotoxicity evaluated (7, 8). Some examples are La(III) complexes with 1,10-phenanthroline-2,9-bis-α-amino acid conjugates (9), coumarines (10), Sm(III) and Gd(III) complexes with acenocoumarol (11), cerium(III) and neodymium(III) complexes with 5-aminooritic acid (12), Gd(III), Dy(III) and Er(III) with dihalo-substituted 8-quinolinol (13).
Meanwhile, recent critical results have shown that new coordination compounds based on active ingredients in traditional Chinese medicines (TCMs) provide a novel approach to develop potential (pro)drugs (14-16) because TCMs have been successfully used to treat diseases among the Chinese for several millennia. A series of active ingredients of TCM, liriodenine/plumbagin/matrine metal-based anticancer compounds, were first reported by our group (17-21). Our further interest focuses on the oxoaporphine metal-based anticancer agents. Oxoglaucine (Figure 1) is an oxoaporphine alkaloid that has been isolated from various plants belonging to different families such as Annonaceae (22), Lauraceae (23), Magnoliaceae (24), Fumariaceae (25), Menispermaceae (26) and Papveraceae (27). The primary screening results revealed that oxoglaucine possessed strong anticancer activity against HCT-8 [50% effective dose (ED50) 2.85 μM] and KB (ED50 5.69 μM) cells (28, 29). Due to its planar aromatic structure, oxoglaucine can intercalate between neighbouring base pairs of a DNA double helix, to which its significant antitumour properties can be primarily attributed. The planar character of oxoglaucine and its N and carbonyl O donors also enable its complexation with metal ions to form metal-based bi-functional compounds with potential synergistic effects on antitumour activity. To explore alkaloid–metal complexes as anticancer agents, we first reported oxoglaucine Au(III), Zn(II), Co(II) and Mn(II) complexes and their in vitro anticancer activity (30). We then proceeded to synthesize and characterize three oxoglaucine–lanthanide complexes to explore the potential of lanthanide complexes as anticancer agents.
Materials and Methods
Materials. All the lanthanide salts and solvents used were of analytical grade. All the materials were used as received without further purification unless noted specifically. Oxoglaucine was synthesized according to a previously reported method (30).
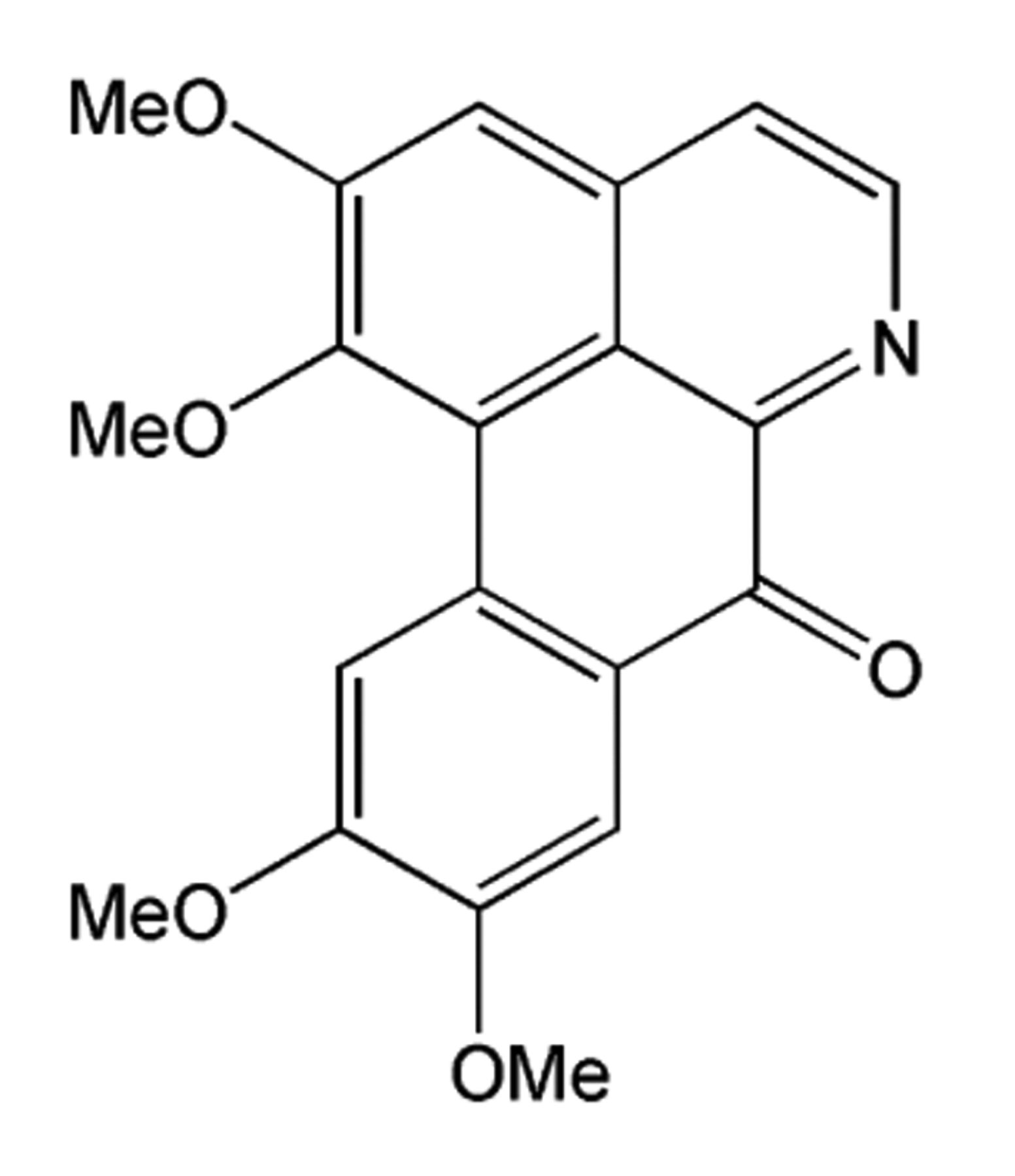
Oxoglaucine.
Measurements. Infrared (IR) spectra were obtained on a PerkinElmer FT-IR Spectrometer (PerkinElmer, Waltham, MA, USA). Elemental analyses (C, H, N) were carried out on a PerkinElmer Series II CHNS/O 2400 elemental Analyzer (PerkinElmer, Waltham, MA, USA). Electrospray Ionization Mass (ESI-MS) spectra were recorded on a Bruker HCT Electrospray Ionization Mass Spectrometer (Bruker Daltonics Inc., Billerica, MA, USA).
Synthesis. [Sm(OG)2(NO3)3]•H2O (1): A mixture of Sm(NO3)3•6H2O (0.044 g, 0.1 mmol), oxoglaucine (0.070 g, 0.2 mmol), 1 ml methanol and 0.25 ml CHCl3 was placed in a thick Pyrex tube (ca. 20 cm long). The mixture was frozen by liquid N2 and evacuated under vacuum and sealed with a torch. It was then heated at 80°C for three days. Red block crystals suitable for X-ray diffraction analysis were collected. Yield: 0.073 g, 69%. analyzed composition (%): C, 45.53; H, 3.41; N, 6.68. Calculated composition for C40H36N5O20Sm: C45.45; H, 3.43; N, 6.63. Main IR (KBr, cm−1): 3426s (−OH), 2943m (Ar–H), 1611m (C=O), 1574m (C=C), 1506m, 1478m, 1383s (NO3−), 1283m (C–O), 1259m, 1008m (C–N). ESI-MS: 823.2 [Sm(OG)2–CH3]+, 550.0 [Sm(OG)+NO3–CH3]+.
[Eu(OG)2(NO3)3]•1.5CH3OH (2): Complex 2 was synthesized in a procedure similar to that of complex 1 except that Sm(NO3)3•6H2O and CHCl3 were replaced by Eu(NO3)3•6H2O and CH2Cl2 respectively. Yellow block crystals suitable for X-ray diffraction analysis were harvested. Yield: 0.065 g, 64%. Analyzed composition (%): C, 45.73; H, 3.75; N, 6.48. Calculated composition for C41.5H40EuN5O20.5: 45.78; H, 3.70; N, 6.43. Main IR (KBr, cm−1): 3422s (−OH), 2945m (Ar–H), 1610m (C=O), 1574m (C=C), 1509m, 1478m, 1385s (NO3−), 1286m (C–O), 1257m, 1008m (C–N). ESI-MS: 824.2 [Eu(OG)2–2CH3]+, 510.9 [Eu(OG)–2CH3+2H2O]+.
[Er(OG)2(NO3)3]•H2O (3): Complex 3 was synthesized in a procedure similar to that of complex 1 except that Sm(NO3)3•6H2O was replaced by Eu(NO3)3•6H2O. Red block crystals suitable for X-ray diffraction analysis were collected. Yield: 0.072 g, 67%. Analyzed composition (%): C, 44.78; H, 3.35; N, 6.58. Calculated composition for C40H36ErN5O20: 44.73; H, 3.38; N, 6.52. Main IR (KBr, cm−1): 3422s (−OH), 2945m (Ar–H), 1608m (C=O), 1574m (C=C), 1509m, 1478m, 1385s (NO3−), 1286m (C–O), 1257m, 1008m (C–N). ESI-MS: 994.3 [Er(OG)2+2NO3]+, 798.2 [Er(OG)+2NO3+2DMSO]+.
Crystal data and refinements for complexes 1-3.
X-Ray crystallography. X-ray crystallography for complexes 1-3 was carried out on a Bruker Apex II Charge-coupled Device (CCD) equipped with graphite monochromated Mo-Kα radiation. The structures were solved using direct methods and refined using the SHELX-97 program (31, 32). The non-hydrogen atoms were located in successive difference Fourier synthesis. The final refinement was performed by full-matrix least-square methods with anisotropic thermal parameters for non-hydrogen atoms on F2. The hydrogen atoms were added theoretically, riding on the concerned atoms, except for the co-crystallized water molecule. The crystallographic data and refinement details of the structure analyses are summarized in Table I.
Cytotoxicity assay in vitro. Liver cancer BEL-7404, human gastric cancer SGC7901, cervical carcinoma HeLa, breast cancer MCF-7, and human lung adenocarcinoma A549 cell lines were obtained from the Shanghai Cell Bank of the Chinese Academy of Sciences. The tumour cells were cultivated in RPMI-1640 medium in suspension containing 10% fetal calf serum, 100 IU/ml penicillin, 100 IU/ml streptomycin at 37°C in an atmosphere humidified with 5% CO2.
In vitro cytotoxicity of quantitative evaluation was determined by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium Bromide (MTT) assay. The study compounds were dissolved in dimethyl sulphoxide (DMSO) and subsequently halved to dilute into five concentrations with RPMI-1640 medium. The solution was stored at 4°C after filtration by a 0.22 μm micropore membrane. BEL-7404 tumour cells in the exponential phase were seeded in a 96-well plate each with 0.19 ml. Study compounds (10 μl) were added to each well after cell cultivation for 12 h. The final concentrations of target compounds were 0.75, 1.5, 3, 6, 12 μg/ml, respectively, in quadruplicates. The final content of DMSO was below 0.5%. Contrast (cells with drug and equal DMSO) and blank (equal drug content without cells) samples were also set up for comparison. After cultivation for 24 h in 37°C, 10 μl MTT [5 mg/ml Phosphate Buffered Saline (PBS)] was each added and cells were cultivated for an additional 4 h. Then the medium solution was removed and 0.15 ml DMSO added to each well. The crystalloids were fully dissolved within 5-min shaking. Absorption values were determined by an enzyme labelling instrument with 550/650 nm double wavelength measurement with the blank group regulated to zero as baseline. The final 50% inhibitory concentration (IC50) values were calculated by the Statistical Product and Service Solutions (SPSS) (33, 34). All the tests were repeated in triplicate.

An Oak Ridge Thermal Ellipsoid Plot (ORTEP) view of [Eu(OG)2(NO3)3]•1.5CH3OH (2) showing atom labeling; thermal ellipsoids are drawn at the 30% probability, and hydrogen atoms as well as one and half co-crystallized methanol molecules have been omitted for clarity.
Results
Synthesis. Oxoglaucine was synthesized by using (+)-boldine as the starting material, as reported in our recent publication (30). Three oxoglaucine lanthanide complexes [Sm(OG)2 (NO3)3]•H2O (1), [Eu(OG)2(NO3)3]•1.5CH3OH (2) and [Er(OG)2(NO3)3]•H2O (3), were prepared in good yields by solvothermal reaction of the corresponding lanthanide salts with the five-membered N,O-chelating ligand oxoglaucine, a route similar to that reported for [Zn(OG)2(H2O)2](NO3)2 (30). The X-ray crystal structures of the three oxoglaucine-lanthanide complexes were determined.
Crystal structure of complexes 1-3. The single-crystal X-ray diffraction analyses of complexes 1-3 revealed that their crystal strutures were very similar, except for the central metal atom and the co-crystallized solvent molecules. In each case, the metal centre is 10 coordinated with the coordination sphere, being completed by two chelating OG and three bidentate NO3− anions. Due to their iso-structural characteristics, we only describe the structure of complex 2; selected bond lengths and angles are reported in Table II.
Selected bond lengths (Å) and angles for complex 2.
As shown in Figure 2, 2 consists of [Eu(OG)2(NO3)3] plus one and half CH3OH molecules in each complex unit. Similarly to EuL1(NO3)3 (L1=2-[2-(1,3-dioxolan-2-yl)quinolin-8-yloxy]-N-benzylacetamide) (35), the central Eu atom is coordinated with 10 donor atoms, six of which belong to three bi-dentate nitrate groups and the remaining four are from the two bi-dentate OG ligands. The whole structure takes the shape of a butterfly–one bi-dentate nitrate ion is on one side of the approximate co-plane formed by two OGs, and the other two bi-dentate nitrate ions are on the other side. The coordination polyhedra around Eu(III) is a distorted bi-capped square antiprism (Figure 2). The bond lengths of Eu–Onitrate and Eu–N are in the range of 2.445~2.593 and 2.576~2.596 Å, respectively, which are comparable to those in EuL1(NO3)3 (Eu–Onitrate 2.452~2.481 Å; Eu–N 2.598) (35). Those for Eu–Ocarbonyl are 2.442(3) and 2.464(3) Å, respectively, which are similar to that of Eu–Ocarbonyl (2.466(5) Å) in tris(6-diphenylmine carbonyl 2-pyridine carboxylato)europium(III) (36). It should be noted that the bond distance of C–Ocarbonyl [1.252(5) and 1.247(5) Å] is shortened in the C=O double bond after the keto O coordinated to Eu(III), in comparison with free OG ligand [C–Ocarbonyl=1.367(4) Å]. However, such a short distance of C–Ocarbonyl in 2 is normal and has been observed in tris(6-diphenylmine carbonyl 2-pyridine carboxylato) europium(III) [C–Ocarbonyl=1.261(11) Å] (36). In the ESI-MS of 2, two major peaks at m/z 824.2 and 510.9 were observed, which could be attributed to two positively-charged species [Eu(OG)2–2CH3]+ and [Eu(OG)–2CH3+ 2H2O]+, accompanying the loss of two methyl groups from the methoxy moieties of the OG ligand under the conditions of the mass spectrometer.
Inhibitory rate (%) of oxoglaucine, complexes 1-3, and corresponding lanthanide salts against five cancer cell lines.
50% Inhibitory concentration (IC50) (μM)a values for complexes 1-3 against five human cancer cell lines.
Similarly to Eu(III) in 2, the coordination geometry of Sm(III) and Er(III) in 1 and 3 could also be described as a distorted bi-capped square antiprism. In the ESI-MS of 1, two major peaks at m/z 823.2 and 550.0 were observed, which could be ascribed to positively-charged species [Sm(OG)2–2CH3]+ and [Sm(OG)+NO3−CH3], and the loss of two methyl groups from the methoxy moieties of the OG ligand under the conditions of the mass spectrometer. Unlike 1 and 2, two major peaks at m/z 994.3 and 798.2 were observed for 3, which could be attributed to two positively-charged species [Er(OG)2(NO3)2]+ and [Er(OG)(NO3)2+2DMSO] without the loss of two methyl groups from the methoxy moieties of the OG ligand under the same conditions. The observed isotopic patterns fit well with the theoretical isotopic distributions.
Cytotoxicity assay in vitro. The in vitro cytotoxicity of oxoglaucine and complexes 1-3 were estimated by MTT assay against liver cancer BEL-7404, human gastric cancer SGC7901, cervical carcinoma HeLa, breast cancer MCF-7, and human lung adenocarcinoma A549 cell lines (with cisplatin as the positive control). As shown in Table III, oxoglaucine (20 μM) exhibited a low inhibitory effect on the tumour cells tested, except for HeLa. The corresponding lanthanide salts against BEL-7404 and HeLa exhibited low activity or no activity, and they were not active even at 100 μM against A549 cells. While at 20 μM, against five tumour cell lines tested, complexes 1-3 were all active, and in most cases, they displayed enhanced cytotoxicity compared to free oxoglaucine and the corresponding lanthanide salts. In order to evaluate the cytotoxicity of complexes 1-3 in detail, we further determined their IC50 values. As tabulated in Table IV, the IC50 values of complexes 1-3 against BEL-7404, SGC7901, HeLa, MCF-7 and A549 are markedly different. Complex 1 was active only against SGC7901 cells, with an IC50 value of 32.1±7.0 μM; complex 2 only active against MCF-7 cells with an IC50 value of 3.2±0.9 μM; and complex 3 was active against both HeLa and MCF-7 cells, with IC50 values of 8.3±3.5 and 1.4±0.4 μM, respectively. In addition, some complexes had lower IC50 values than cisplatin, such as complexes 2 and 3 to MCF-7.
Discussion
In our study, the three lanthanide-OG complexes are iso-structural with a distorted bi-capped square antiprism geometry and chelate two OG ligands. The ESI-MS results of complexes 1-3 suggest that two forms of species exist with metal: ligand molar ratios of 1:2 and 1:1, both of which still possess a planar OG moiety resulting in their high cytotoxicity. From the inhibitory rate of complexes 1-3 on five cell lines, it was found that in most cases, they displayed enhanced cytotoxicity compared to free oxoglaucine and the corresponding lanthanide salts. Against MCF-7 cells, complexes 2 and 3 performed better than cisplatin. Even though complexes 1-3 possess similar structures, these OG–lanthanide complexes have different activity profiles against the tested cell lines, which is difficult to explain. In fact, except for cisplatin, there is relatively little mechanistic information on how metal anticancer drugs function, but it is clear that different metal ions can act through different routes that lead to different cellular responses (37).
In conclusion, three new mononuclear lanthanide complexes 1-3 with oxoglaucine have been synthesized and fully characterized. The results of in vitro cytotoxicity assay of complexes 1-3 against BEL-7404, SGC7901, HeLa, MCF-7 and A549 cells indicated that their effect on tumour cells were markedly different: complex 1 was active only against SGC7901 cells, complex 2 only against MCF-7 cells and complex 3 against HeLa and MCF-7 cells. It seems that these OG–metal complexes were selectively active against certain cell lines. In some cases, they exhibited significant enhanced antitumor activity compared with that of oxoglaucine and its corresponding metal salts.
Acknowledgements
This work was supported by the National Basic Research Program of China (nos. 2010CB534911, 2012CB723501) and the Natural Science Foundation of Guangxi Province (nos. 2012GXNSFDA053005, 2010GXNSFF013001), and IRT1225 as well as the BAGUI Scholar Program of Guangxi of China.
Footnotes
-
↵Conflicts Interest
None.
-
Supplementary Material
Crystallographic data for the structural analysis have been deposited with the Cambridge Crystallographic Data Centre, CCDC no. 863841, 863839, 808337 for complexes 1-3, respectively. The data can be obtained free of charge via http://www.ccdc.cam.ac.uk, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, U.K. Fax: +44 1223336033, or e-mail: deposit{at}ccdc.cam.ac.uk
- Received August 19, 2013.
- Revision received November 28, 2013.
- Accepted November 29, 2013.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.