


Abstract
The frequent development of cellular resistance to cisplatin in cancer patients is a serious limitation for clinical drug therapy. However, cisplatin resistance is incompletely understood. We have shown that cisplatin-resistant A2780 ovarian cancer cells (A2780cis) can efficiently be eliminated by liposomal cisplatin, which displayed similar cytotoxicity towards both A2780 and A2780cis cells. This may, at least in part, be related to a higher intracellular accumulation of the drug within the resistant cells after liposomal entry. However, the superior cytotoxicity of the liposomal drug was not reflected by DNA platination. This suggests a more complex mode of action of liposomal cisplatin, most likely affecting different signaling pathways. To gain insight into the resistance gene signature, a whole-genome gene expression analysis was performed for A2780cis cells, untreated or treated with half-minimal inhibitory concentration (IC50) of free and liposomal cisplatin. Strong differences in the functional networks affected by free and liposomal cisplatin became evident. p53 was identified as a key factor directing differences in the apoptotic processes. While free cisplatin induced the intrinsic pathway of apoptosis, liposomal cisplatin induced expression of genes of DNA damage pathways and of the extrinsic pathway of apoptosis. These predictions from gene expression data were confirmed at the protein and function level. This sheds new light on liposomal drug carrier approaches in cancer and suggests liposomal cisplatin as a promising strategy for the treatment of cisplatin-resistant ovarian carcinoma.
Liposome-based formulations of cytostatic agents hold great promise in cancer therapy. The tendency of liposomal carriers to accumulate in solid tumour tissues based on the Enhanced Permeability and Retention (EPR) effect (1) helps to modify drugs pharmacokinetics, enhance the drug index and reduce toxic side-effects, which are often limiting for therapy (2).
Cisplatin, an agent for standard treatment of head and neck or ovarian malignancies, has also been produced in a liposomal formulation and reached clinical development in phase III (3). Liposomal cisplatin reduces oto- and nephrotoxicity and in combination with other drugs leads to a higher response rate, which is essential, considering chemoresistance after recurring cycles of treatment is one of the greatest problems in platinum-based chemotherapy (4). However, the question of whether liposomal cisplatin functionally interferes with chemoresistance remains open.
In a previous study, we showed that liposomal cisplatin can overcome chemoresistance in ovarian tumour cells, most likely by by-passing uptake transporter restrictions e.g. down-regulation of the copper transporter-1 (CTR-1) (5). Although liposomes serve as effective carriers and raise intracellular platinum levels in resistant cells, DNA-platination data contradicted the fact that ‘re-acquired’ sensitivity was a simple concentration effect.
Recently, a transcriptome analysis with subsequent process network identification of cisplatin-resistant A2780 ovarian tumour cells in comparison with sensitive cells revealed different gene expression induced by liposomal cisplatin in comparison to the free drug (6).
These data will be revisited here to underline our current studies directed at the expression and functional activity of key players in apoptosis, such as caspase-3, -8 and -9, to illustrate the differences in cytotoxicity of free vs. liposomal cisplatin and as a key to understanding the potential of liposomes to overcome resistance.
Materials and Methods
Liposome preparation. Cisplatin liposomes were prepared from soy phosphatidylcholine (SPC), cholesterol and 1,2-distearoyl-snglycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (ammonium salt) (mPEG-PE) in a 65:30:5 molar ratio by hydration technique as described (5). Briefly, a chloroform solution of the lipids was evaporated and hydrated with 8 mg/ml cisplatin in a 0.9% saline solution. Size homogenisation was applied by ultrasonication and cisplatin excess removed by gel permeation chromatography (Sephadex G-50®; Sigma-Aldrich Chemie, Steinheim, Germany).
Liposomes were characterised by determining particle size and ζ-potential (Zetatrac, Microtrac), as well as phospholipid and cisplatin concentration by flameless Atomic Absorption Spectroscopy (fAAS) as indicated elsewhere (5). Stability was proven by 6-carboxyfluorescein (6-CF) leakage at different pH levels and the toxicity of empty liposomes was assessed by 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium-bromide (MTT) assay as described below.
SPC was purchased from Lipoid AG, Ludwigshafen, Germany, mPEG-PE from Genzyme Pharmaceuticals, Neu-Isenburg, Germany and cholesterol and cisplatin were from Sigma-Aldrich Chemie.
Cell lines. The human ovarian carcinoma cell line A2780 and cisplatin-resistant A2780cis cells were obtained by the European Collection of Cell Cultures, Salisbury, UK. They were cultured in RPMI-1640 medium with 10% fetal calf serum (FCS), 1.5% L-glutamine and 1% penicillin/streptomycin supplement (PAN Biotech, Aidenbach, Germany). In order to maintain chemoresistance in A2780cis cells, the cells were used for only eight passages from working stock.
Cellular cisplatin accumulation. The platinum uptake by both cell lines was analysed by fAAS for different incubation times between 0.5 and 24 h and related to total protein amounts (5).
Cisplatin toxicity. The toxicity of free and liposomal cisplatin in A2780 and A2780cis cells was detected by the MTT assay in which the UV absorption of formazan from MTT correlates directly with cell vitality.
20,000 cells were seeded in 100 μl/well in 96-well plates and allowed to grow overnight. Cisplatin was then added at concentrations from 10−3.5 mol/l to 10−8.5 mol/l, covering the typical IC50 range of A2780 and A2780cis cells known from previous experiments. Empty liposomes were tested for toxicity in a total lipid concentration from 10-1.5 mol/l to 10-5.5 mol/l. After 24, 48 and 72 h incubation (37°C, 5% CO2), the UV/VIS absorption of each sample was measured.
To evaluate the kind of apoptosis pathway contributing to cell toxicity of cisplatin, different inhibitors of p53-dependent caspases were added. Since caspase-8 is pivotal for the extrinsic apoptosis pathway, and caspase 9 reflects the mitochondrial intrinsic apoptotic activity (7), the effect of the caspase-8 inhibitor Z-IETD-FMK and caspase-9 inhibitor Z-LEHD-FMK, compared to the caspase-3 inhibitor Z-DEVD-FMK (all purchased from R&D Systems, Abingdon, UK) on cytotoxicity of free and liposomal cisplatin was investigated. Therefore, an aliquot of the cells was preincubated with 50 μmol/l caspase-3, -8 or -9 inhibitor for 3 h before cisplatin treatment. UV/VIS absorption was measured 72 h after incubation.
DNA-platination. Cell preparation and quantification of DNA-bound platinum were performed as described in (6).
Gene array analysis. The revisited transcriptome data were acquired using an Agilent Microarray Scanner and its extraction software and furthermore processed by GeneGO, as described elsewhere (6).
Apoptosis array–proteome profiler™. Cisplatin-resistant cells (A2780cis) were treated with 2 μmol/l of either free or liposomal cisplatin, or empty liposomes in a total lipid concentration corresponding to 2 mmol/l cisplatin liposomes for 72 h. After 72 h, cell lysates were prepared and apoptosis induction was assessed by an apoptosis antibody-array kit following the manufacturer's instructions (R&D Systems). Untreated cells served as background control.
Here, apoptosis proteins are bound to immobilised monoclonal antibodies (mAbs), spotted on a membrane, and are linked with biotinylated secondary mAbs in the manner of an enzyme-linked immunosorbent assay. The detection was performed by horseradish peroxidase-conjugated streptavidin and chemiluminescence measurement (ChemiDoc® XRS+; Bio-Rad Laboratories GmbH, Munich, Germany). Results are shown as the ratio of the test to the control.
Expression of caspases. The expression of the caspases-8 and -9 was examined by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and western blotting (Mini Trans-Blot® Cell; Bio-Rad Laboratories GmbH). Therefore, lysates were prepared from A2780 and A2780cis cells incubated with IC50 of liposomal cisplatin and free drug over 72 h (37°C, 5% CO2).
For detection, we used a mouse antibody to caspase-8/-9 (R&D Systems), which was bound to a complementing HRP-conjugated anti-mouse secondary antibody. Amplification was conducted via the Luminol reaction. ECL solution, Precision Plus Standard and Streptactin were purchased from Bio-Rad.
Results and Discussion
Enhanced uptake of liposomal vs. free cisplatin by A2780cis cells. Reduced intracellular Pt levels are a common reason for resistance to cisplatin, e.g. by down-regulation of CTR-1. To overcome this restriction we aimed to prepare cisplatin liposomes for an endocytotic cell entry (8) avoiding uptake restrictions. The prepared liposomes had a mean size of 113 nm and cisplatin concentration of 520 μmol/l, representing a mean entrapment efficiency of 4.6% and sufficient loading stability.
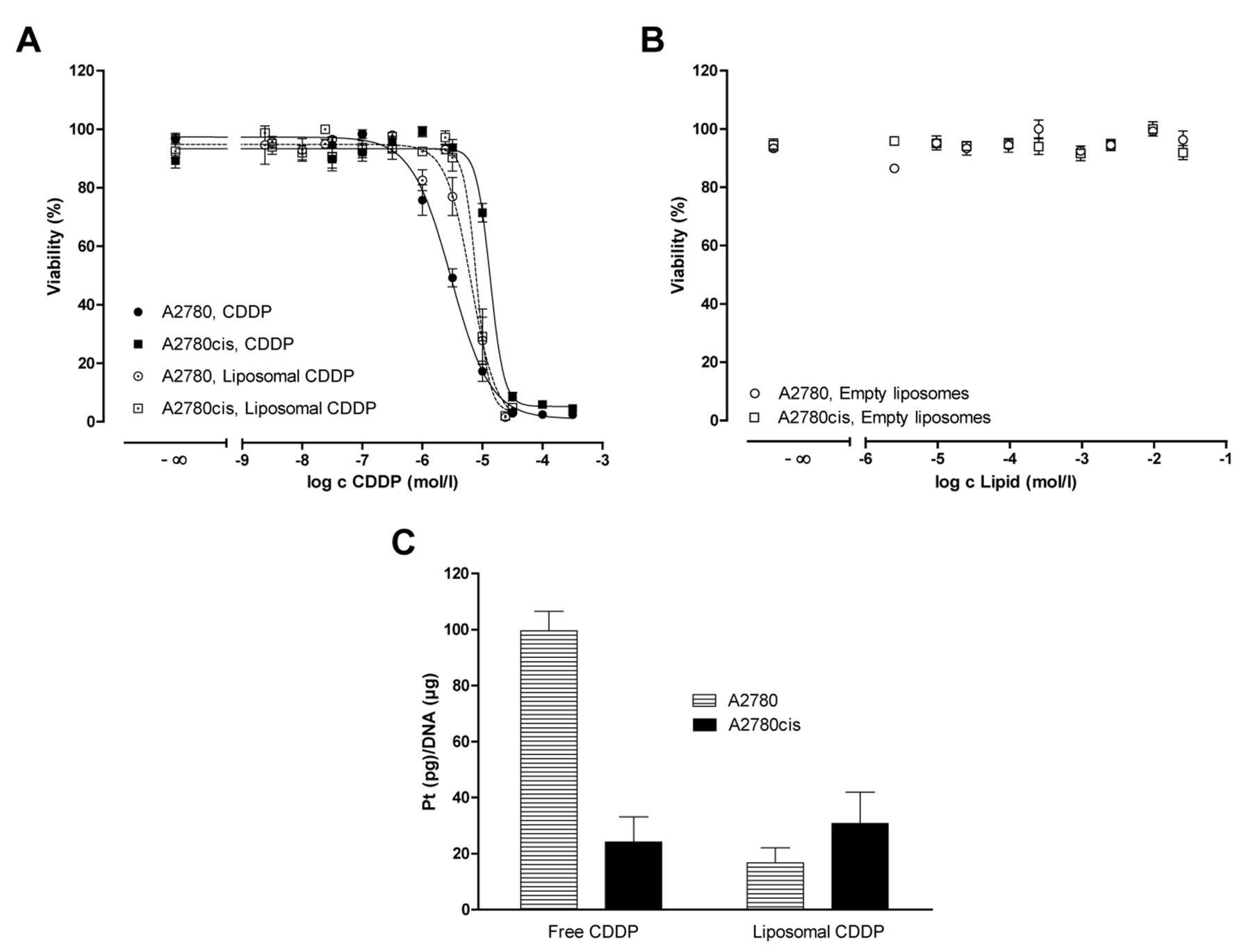
Considering the uptake of cisplatin in A2780 and A2780cis cells for a 24-h period (Figure 1), a crucially reduced uptake of free cisplatin into A2780cis cells is evident, reflecting the resistant behaviour of the cells. The much higher cisplatin concentration in A2780 cells impressively reflects the balance of influx and efflux based on transporters and their impact on resistance (9).
In contrast, liposomal cisplatin was accumulated in both cell lines to a similar extent (~40 ng/μg after 24 h), which clearly exceeded the level of the free drug in the resistant cells (Figure 1). This supports our postulation on the value of endocytotic cell uptake and confirms the carrier potential of liposomes.

Cellular uptake of free vs. liposomal cisplatin (CDDP) in A2780 and A2780cis cells. CDDP concentration was 20 μmol/l in each experiment. Data points are means±SD of three experiments performed in triplicates.
Overcoming of cisplatin resistance by liposomal cisplatin. Chemoresistance is clearly reflected by the findings of the MTT assay shown in Figure 2A, where the free drug had an IC50 of 3.00 μmol/l in A2780 cells and 13.39 μmol/l in A2780cis cells, leading to a resistance factor (Rf=IC50 A2780cis/IC50 A2780) of 4.5.
In contrast, liposomal cisplatin demonstrates higher cytotoxicity than the free drug against A2780cis cells (IC50=8.01 μmol/l; Rf=1.2), although plain liposomes do not possess any cytotoxicity per se (Figure 2B).
DNA binding of free and liposomal cisplatin. The strong difference in DNA-platination levels by free cisplatin in A2780 and A2780cis cells (Figure 2C) clearly reflects the differences in cell uptake and cytotoxicity in both cell lines. However, despite higher uptake and activity of liposomal cisplatin in A2780cis cells, the level of platination is probably not an indicator of increased activity, with a rather low value of ~25 pg/μg, comparable to the free drug in A2780cis cells. Obviously, liposomal cisplatin has a different mode of action of cytotoxicity.
Differences in transcriptome after cisplatin treatment. Since functional assays do not provide mechanistic insights into resistance phenomena, a gene array analysis was performed. All details on de-regulated genes and their functional analysis are given elsewhere (6). In general, A2780cis cells treated with IC50 of either free or liposomal cisplatin strictly differ in their genetic response. As a general overview, a GeneGO process network analysis is given (Figure 3), which illustrates that the 10 most de-regulated pathways are different in A2780cis cells treated with free cisplatin from these treated with liposomal cisplatin. Furthermore, p53 was identified as an essential factor for mediating transcriptional responses after exposure to free and to liposomal cisplatin. While the free drug induced expression of essential genes of the intrinsic apoptosis pathway [Bcl-2-associated × protein (BAX), BH3 interacting-domain death agonist (BID), caspase-9 gene (CASP9)], most likely through activation of p38 mitogen-activated protein kinases (p38MAPK), liposomal cisplatin induced expression of genes of the extrinsic pathway of apoptosis [tumour necrosis factor receptor superfamily member 10b (TNFRSF10B), tumour necrosis factor ligand superfamily member 7 (TNFSF7)]. These findings illustrate that liposomal cell entry of cisplatin has important consequences for the affected cellular targets and apoptosis in comparison to the free drug.
Expression of apoptosis-related proteins. To investigate whether the genetic findings on different apoptosis induction by free and liposomal cisplatin are verified on the protein level, an antibody proteome profiling array was applied. As shown in Figure 4, both liposomal and free cisplatin induced an up-regulation of cleaved caspase-3 protein, which indicates cytotoxic and apoptotic effects, since caspase-3 is a downstream effector of intrinsic and extrinsic pathways of apoptosis. These results are specific for cisplatin, as empty liposomes did not significantly induce cleaved caspase-3.
Interestingly, free cisplatin caused significant up-regulation of cytochrome c (~30%) and BAX (~10%) proteins (Figure 4), which are involved in intrinsic (mitochondrial) apoptosis (10). In contrast, liposomal cisplatin caused a down-regulation of these components of the intrinsic apoptosis pathway. On the other hand, apoptosis-stimulating fragment (FAS) and FAS-associated protein with Death Domain (FADD), as indicators of early apoptosis and sensors for extracellular stimuli and downstream signaling (11), were not induced by the free drug (Figure 4), but displayed higher expression (FAS) after liposomal cisplatin treatment. These findings show that treatment of A2780cis cells with free cisplatin results in activation of the intrinsic pathway of apoptosis, while liposomal cisplatin induces cell toxicity by extrinsic apoptosis activation.
Expression of participating caspases. To gain further insight into the apoptotic processes and differences thereof, we first focused on expression levels of caspases in A2780cis cells after free and liposomal cisplatin treatment.
As for the extrinsic pathway a higher processing of the 55/53 kDa inactive caspase 8 to the 42-kDa cleaved form was found after treatment with liposomal CDDP (Figure 5A).
Caspase 9 precursor of 43 kDa was almost fully processed to the cleaved form with a major fraction of the p35 effector (Figure 5B). Thereby, cells incubated with free drug exhibited a higher protein expression of the caspase 9 active form of 35 kDa. They even displayed a higher amount of caspase 9 complexed with apoptotic protease activating factor-1 (APAF-1), which is an essential requirement for the formation of the apoptosome and intrinsic apoptosis.

Cell viability data from cytotoxicity assays (20,000 cells/well). A: Comparison of the cytotoxicity of free vs. liposomal cisplatin (CDDP) after 72 h in A2780 or A2780cis cells. Data are means±SEM of three experiments performed in triplicates. B: After 72 h, no toxic effect of empty liposomes was evident. C: DNA-platination of A2780 or A2780cis cells after treatment with 20 mmol/l free or liposomal CDDP for 24 h. Data are means of at least three independent experiments (n=3 in triplicates±SD).
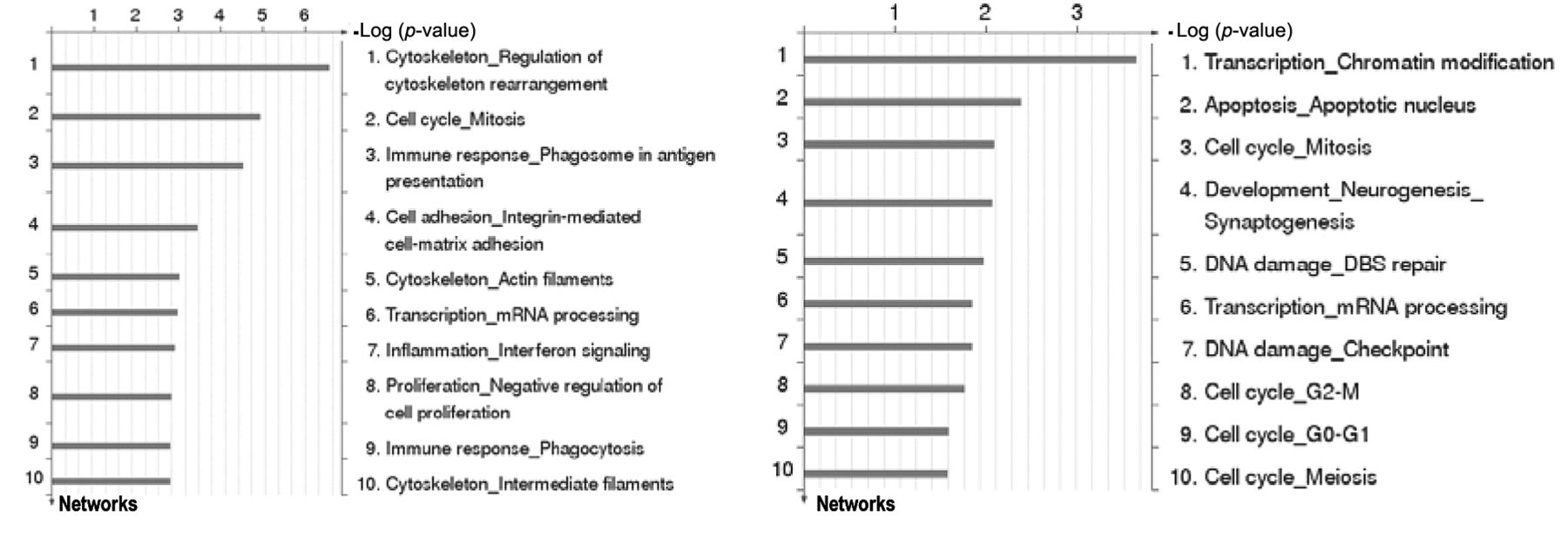
Identification of significant GeneGo process networks in A2780 and cisplatin-resistant A2780cis ovarian cancer cells after exposure to free cisplatin at half minimal inhibitory concentration (IC50).

Effect of treatment with free cisplatin (CDDP), liposomal CDDP and empty liposomes on expression of apoptotic proteins in A2780cis cells assessed by Human Apoptosis Array Kit-proteome profiler™ (right). Cells were treated for 72 h with either free or liposomal CDDP. Results are mean of duplicates, which individually differed by less than 5.4% RSD. Protein deregulations (expressed as ratios, left) are significant according to one-way ANOVA (p<0.001). Bcl-2-associated death promoter (BAD), Bcl-2-associated X protein (BAX), FAS-associated protein with Death Domain (FADD), apoptosis-stimulating fragment (FAS), 27, 60 and 70 kDa heat shock proteins (HSP27, HSP60, HSP70).

A2780cis cells after 72 h incubation with half minimal inhibitory concentration of free drug vs. liposomal cisplatin (CDDP). Immunoblotting for caspase-8 (A) and caspase-9 (B).Percentage increase in IC50 of free and liposomal CDDP in A2780cis cells assessed by cytotoxicity assays (20,000 cells/well, 72 h) after preincubation with different caspase inhibitors (C). Data are the mean±SD of two experiments performed in triplicates.
Thus, A2780cis cells treated with free cisplatin have a caspase protein profile which is characteristic for the intrinsic pathway, while cells incubated with liposomal cisplatin are more likely to process the extrinsic precursor caspase 8.
Selective activation of different apoptosis pathways. To investigate the functional involvement of different caspases in the cytotoxic effect of free and liposomal cisplatin and to elucidate differences in the apoptotic pathways, MTT assays of A2780cis cells in the presence of different caspase inhibitors were performed. If apoptosis is inhibited by caspase inhibitors, more cisplatin will be needed to achieve a 50% cell kill, i.e. the IC50 will increase.
Inhibition of downstream effector caspase 3, which is part of the extrinsic and intrinsic pathway (7), led to a lower IC50 for liposomal cisplatin than the free drug (Figure 5C). With caspase-8 inhibitor, a significantly higher concentration of liposomal cisplatin was needed than free drug. This demonstrates that liposomal cisplatin activates the extrinsic pathway of apoptosis in A2780cis cells. In contrast, the IC50 for free cisplatin in cells with caspase-9 inhibition was much higher than that for liposomal cisplatin. This result shows the strong activation of the intrinsic pathway by free drug in contrast to liposomal drug.
Taken together our results show a selective activation of the extrinsic pathway of apoptosis by liposomal cisplatin in cisplatin-resistant A2780cis ovarian cancer cells and provide an explanation as to how liposomal formulation can overcome cisplatin resistance of A2780cis ovarian cancer cells.
Conclusion
Liposomes have intensively been investigated with respect to their capacity to target cytostatic agents to solid tumours. Thereby, liposomes have most often been considered as passive carriers in these approaches. Here, we provide evidence that a liposomal drug transport into cells might have greater consequences for directing the drug activity beyond its simple transport function. We show here by gene array, protein expression and functional data that liposomal cisplatin induces programmed cell death via the extrinsic pathway of apoptosis, which is in contrast to the drug taken-up in free form. This emphasises the importance of cellular uptake routes for directing the intracellular trafficking of the drug, balancing the effects by different intracellular targets. This is clearly the reason why liposomal cisplatin acts differently from the free drug and thus overcomes cellular resistance in A2780cis cells. These findings shed new light on liposomal drug carrier approaches in cancer and suggest liposomal cisplatin as a promising strategy for the treatment of cisplatin-resistant ovarian carcinomas.
- Received August 20, 2013.
- Revision received November 27, 2013.
- Accepted November 28, 2013.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}