


Abstract
The present study investigated the effect of the H2 antagonist cimetidine on the pharmacokinetics of a multi-targeted receptor tyrosine kinase (RTK) inhibitor, sunitinib, in Sprague-Dawley (SD) rats and Eisai hyperbilirubinemic mutant rats (EHBR) lacking the efflux transporter, ATP-binding cassette C2 protein (ABCC2). Rats received an intraperitoneal injection of cimetidine (10 mg/kg) once a day for three days. On day 4, sunitinib (3 mg/kg) was administered intravenously 30 min after the final injection of cimetidine or saline to SD rats. Disappearance of sunitinib from plasma was significantly delayed by cimetidine. The pharmacokinetic parameter of sunitinib, systemic clearance (CLSYS), was significantly reduced and the half-life was significantly prolonged, with no change in the volume of distribution at steady-state (VSS). When the effect of cimetidine on the biliary excretion of sunitinib at steady-state condition was investigated in SD rats, cimetidine had no effect on some transporter-mediated biliary excretion of sunitinib. Furthermore, the contribution of ABCC2 to the biliary excretion of sunitinib was also examined in SD rats and EHBR. The biliary clearance of sunitinib was significantly lower in EHBR, but the biliary excretion rate of EHBR was not different from that of SD rats, and the contribution of biliary excretion to systemic elimination was small, suggesting that sunitinib is mainly eliminated by cytochrome P450 3A4 (CYP3A4)-mediated metabolism and is not excreted into the bile via ABCC2. These findings indicate that co-administration of cimetidine alters the pharmacokinetics of sunitinib probably due to inhibition of CYP3A4, suggesting the possibility that cimetidine should be used carefully for patients with cancer being treated with sunitinib therapy.
- Biliary excretion
- cimetidine
- drug interaction
- pharmacokinetics
- multidrug resistance-associated protein-2 (Mrp2)
- sunitinib
- (Abcc2)
It is well-known that the ATP-binding cassette (ABC) efflux pumps, ABCB1 (P-glycoprotein), ABCG2 (breast cancer resistance protein, BCRP) and ABCC2 (multidrug-resistance-associated protein 2, MRP2), are the three major canalicular efflux transporters located on the canalicular membrane of hepatocytes (1). These efflux transporters play an important role in the hepatobiliary excretion of various drugs and have a protective function against endogenous and exogenous toxic compounds.
According to “Additional Drug Information” of each pharmaceutical company, many oral small-molecule multi-targeted drugs widely used in cancer therapy have almost the same physicochemical properties such as basic drugs, i.e. relatively high hydrophobicity [partition coefficient (log P) >2] and high protein-binding potency (95%). Because of the pharmacokinetic characteristics of these drugs, they are primarily metabolized by cytochrome P450 3A4 (CYP3A4) and partly excreted into bile. Among them, sunitinib, a multi-targeted receptor tyrosine kinase inhibitor, is a potent inhibitor of vascular endothelial growth factor receptors (VEGFR)-1, -2, and -3, which are involved in angiogenesis, and platelet-derived growth factor receptors (PDGFRs) (2). Sunitinib is used for the treatment of advanced or metastatic renal cell carcinoma and imatinib-resistant gastrointestinal stromal tumors, and is extensively metabolized by CYP3A4, resulting in the formation of an active metabolite, N-desethyl sunitinib (3, 4). It is reported that sunitinib and N-desethyl sunitinib are substrates of P-glycoprotein and ABCG2, but not substrates of ABCC2 (5-7). Based on these findings, sunitinib may be actively excreted into bile via these efflux transporters. However, there are no reports supporting the contribution of these efflux transporters which are expressed in the canalicular membranes of hepatocytes to the biliary excretion of sunitinib. In addition, sunitinib is clinically used as an oral formulation and is thus often co-administered with various drugs, including anticancer drugs. Therefore, there is a possibility that drug interaction between sunitinib and concomitant drugs, such as substrates, inducers and inhibitors of CYP3A4 and P-glycoprotein, may occur.
It is well-known that the H2 antagonist cimetidine irreversibly inhibits CYP3A4 and is mainly excreted into the urine via the organic cation transport system. Cimetidine is a substrate of P-glycoprotein, like sunitinib (8-10). Cimetidine inhibits angiogenesis and suppresses tumor growth (11), resulting in considerably enhancing survival rates in patients with gastric and colorectal cancer (12-14). Interestingly, it is reported that cimetidine has survival benefit over patients with renal cell carcinoma, or advanced or metastatic renal cell cancer (15-17). There is therefore a possibility that sunitinib may also be co-administered with cimetidine for these patients.
Considering that a majority of the small-molecule targeted anticancer drugs are weak bases, when used concomitantly with gastric acid secretion inhibitors, including H2 antagonists, their gastrointestinal absorption may be altered by increasing gastric pH. Therefore, therapy with sunitinib plus cimetidine may alter the plasma concentration of sunitinib by interfering with its gastrointestinal absorption, in addition to the inhibition of CYP3A4-mediated metabolism and P-glycoprotein- and/or ABCG2-mediated biliary excretion. However, to our knowledge, there is no information regarding drug interactions between sunitinib and cimetidine.
The aim of the present study was to create a guideline for the safe use of cimetidine for patients with cancer who are scheduled to receive sunitinib therapy. To investigate whether cimetidine alters the systemic pharmacokinetics of sunitinib in rats, sunitinib was administered intravenously to exclude the effect of cimetidine on the absorption process. We also investigated the contribution of the efflux transporters P-glycoprotein and Abcc2 to the biliary excretion of sunitinib using Eisai hyperbilirubinemic mutant rats (EHBR) which have a hereditary deficiency in Abcc2 (18).
Materials and Methods
Chemicals. Sunitinib malate was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). Imatinib mesylate, which was used as an internal standard for the measurement of concentrations of sunitinib in plasma and bile, was purchased from BioVision (Milpitas, CA, USA). Cimetidine for injection (Tagamet®) was purchased from Dainippon Sumitomo Pharma (Osaka, Japan). All other chemicals were commercially available and were of the highest purity available. All reagents were used without further purification. For animal experiments, sunitinib was dissolved in citric acid buffer (pH 4.7) at a concentration of 3 mg/ml.
Animals and experiments. Male Sprague-Dawley (SD) rats (age=9 weeks; body weight=342 to 360 g) and male EHBR (body weight=340 to 365 g) of the same age as the SD rats were obtained from Japan SLC Inc. (Hamamatsu, Japan). The rats were housed under controlled environmental conditions (temperature of 23±1°C and humidity of 55±5%) with commercial diet and water freely available. All animal experiments were carried out in accordance with the guidelines of Nagoya University for the care and use of laboratory animals (024-022).
To investigate the effect of cimetidine on the pharmacokinetics of sunitinib, cimetidine (10 mg/kg) was administered intraperitoneally to rats once a day for three days. On day 4, under anesthesia by intraperitoneal injection of sodium pentobarbital (25 mg/kg of body weight), the rats were cannulated with polyethylene tubes into the right jugular vein for drug administration and blood sampling. After awakening, sunitinib (3 mg/kg) was administered 30 min after a final administration of cimetidine (10 mg/kg). Control rats received isotonic saline (0.2 ml/100 g). Blood samples (0.2 ml) were collected at designated time intervals (15, 30, 45 min and 1, 2, 4, 6, 8 and 10 h after injection of sunitinib). Plasma samples were obtained from the blood samples by centrifugation at 3,000 ×g for 10 min at 4°C and stored at −70°C until analysis.
For biliary excretion experiments using SD rats and EHBR, rats under anesthesia by intraperitoneal injection of sodium pentobarbital (25 mg/kg of body weight) were cannulated in the right jugular vein, left carotid artery and bile duct for drug administration, blood sampling and bile collection, respectively. After surgical preparation, rats received a bolus injection of sunitinib at a loading dose of 0.2 mg/kg, followed by a constant-rate infusion of a saline solution at a dose of 111 μg/h/kg with a Harvard infusion pump (PHD 2000; Harvard, South Natick, MA, USA). The loading and maintenance doses of sunitinib were determined by using pharmacokinetic parameters obtained from the single intravenous injection experiment. After a 60-min infusion, bile samples were collected at 30-min intervals within 60 min in SD rats and EHBR. After a 120-min infusion, cimetidine (10 mg/kg) was administered intravenously in order to investigate the effect of cimetidine on the biliary excretion of sunitinib in SD rats. Bile samples were collected at 30-min intervals between 120 and 180 min. Blood samples (0.2 ml) were collected at midpoints of the bile collection periods (75, 105, 135 and 165 min after the infusion was started). Plasma samples were obtained by immediate centrifugation of blood samples. The volume of bile samples was measured gravimetrically, with the specific gravity assumed to be 1.0. Plasma and bile samples were stored at −70°C until analysis. This experiment was carried out under anesthesia with pentobarbital, and the body temperature of the animals was maintained at 37°C throughout the experiments with a heat lamp.
Drug analysis. Concentrations of sunitinib in plasma and bile were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (19) with slight modification. Sunitinib standard solution was prepared at a concentration of 1 mg/ml in acetonitrile and was diluted with methanol to each working standard solution at concentrations ranging from 0.1 to 10 μg/ml. A matrix-matched calibration curve for sunitinib was used for sunitinib standards. Twenty microliters of plasma or bile sample were mixed with 380 μl of distilled water, 800 μl of methanol and 50 μl of acetonitrile containing the internal standard imatinib at a concentration of 10 μg/ml. These mixtures were passed through a solid-phase extraction column, Captiva NDripid (Agilent Technologies, CA, USA). The samples were then injected into LC-MS/MS (Agilent Technologies 6430 series).
Optimized mass spectrometry parameters for determination of sunitinib and imatinib.
Chromatographic separation by LC-MS/MS was performed by a ZORBAX SB-Aq column (2.1×50 mm, 1.8 μm), using a gradient elution mode of water containing 0.1% formic acid (mobile phase A) and of acetonitrile containing 0.1% formic acid (mobile phase B). Linear gradient elution employ the mobile phases at a ratio of A:B as follows: 0 min, 95:5; 1 min 95:5; 4 min, 20:80; 6 min, 20:80. Equilibration time was 3 min. Total flow rate was 0.2 ml/min. Ionization of sunitinib was performed by positive electrospray ionization mode. The main parameter settings of the quantitative technique are shown in Table I. Acquisition was performed using the multiple reaction monitoring (MRM) mode, monitoring two transitions per compound (one for quantification and the other for confirmation) and one for the internal standard as shown in Table I.
These assays were shown to be linear for the concentrations tested, with a correlation coefficient of 0.998. The within- and between-day coefficients of variation for these assays were less than 14%. The detection and quantification limits of sunitinib were 1 and 3 ng/ml, respectively.
Data analysis. Plasma concentration–time data for sunitinib in each rat after a single intravenous administration were analyzed individually using a non-compartmental model. The area under the plasma concentration–time curve (AUC) and the area under the first-moment curve (AUMC) were calculated by the trapezoidal method until the last measurable concentration in plasma and were then extrapolated to infinity. The systemic clearance (CLSYS) was calculated as dose/AUC. The mean residence time (MRT) was calculated as AUMC/AUC. The steady-state volume of distribution (VSS) was calculated as CLSYS × MRT.
For biliary clearance experiments, the biliary clearance based on the concentration in plasma (CLBILE) was calculated by dividing the biliary excretion rate by the steady-state concentration of sunitinib in plasma (CSS) determined for that collection period.
Statistical analysis. Results are expressed as the mean±standard deviation (SD) for the indicated numbers of experiments. Statistical comparisons were assessed by Student's t-test. p-Values less than 0.05 were considered statistically significant. Stat View software (Abacus Concepts Inc., Berkeley, CA, USA) was used for the analysis.
Pharmacokinetic parameters of sunitinib after a single intravenous administration in cimetidine-treated and untreated SD rats.
Results
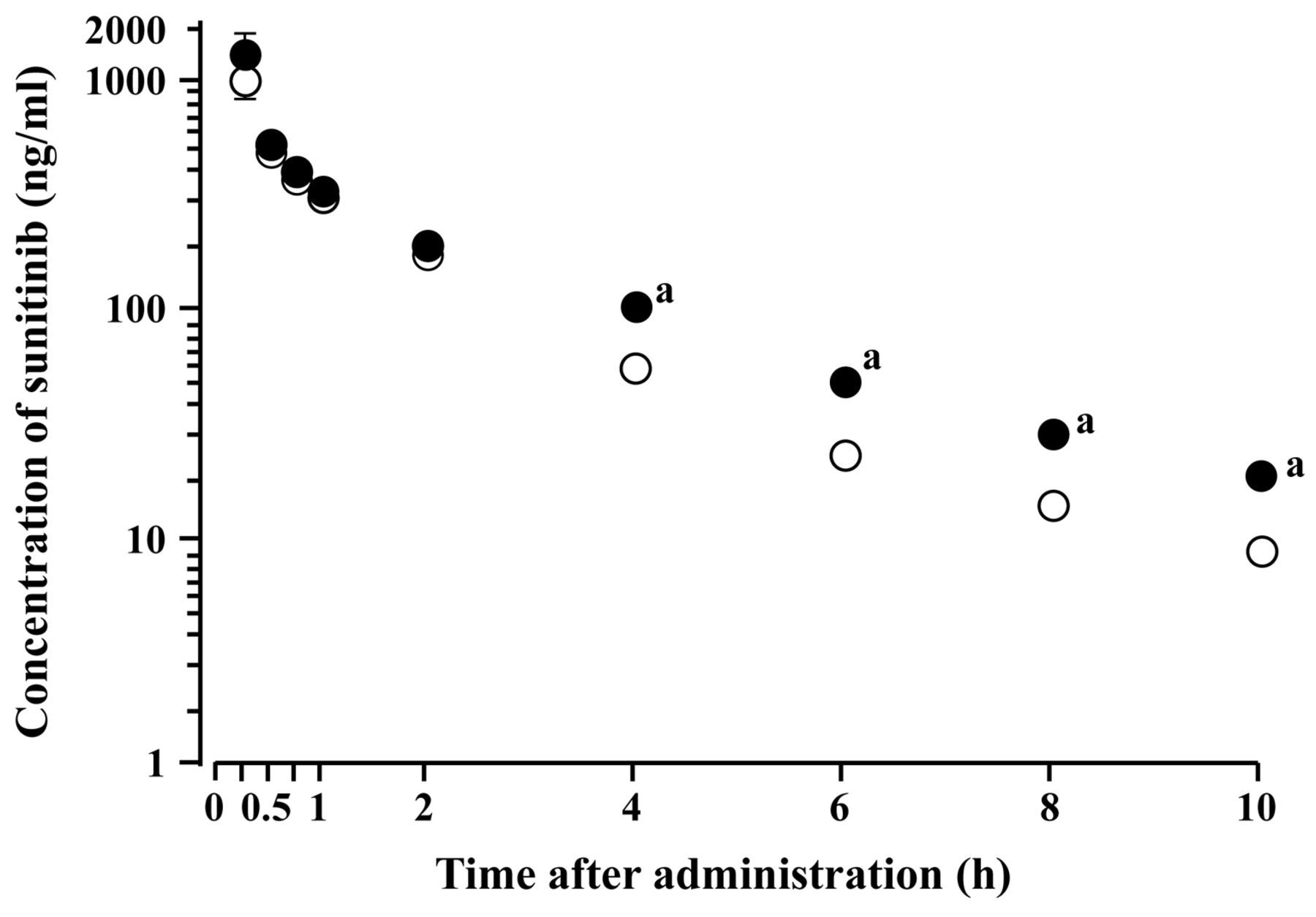
Effect of cimetidine on pharmacokinetics of sunitinib. The effect of treatment with cimetidine on the pharmacokinetics of sunitinib was examined in SD rats. Semi-logarithmic plots of plasma concentration–time data for sunitinib after a single intravenous injection of sunitinib in cimetidine-treated and untreated (control) rats are illustrated in Figure 1. As shown in Figure 1, sunitinib was biphasically eliminated from plasma after intravenous injection. Disappearance of sunitinib from plasma was significantly delayed in cimetidine-treated rats.
The corresponding pharmacokinetic parameters of sunitinib are summarized in Table II. Treatment with cimetidine significantly reduced the CLSYS of sunitinib and prolonged the terminal half-life (t1/2). However, no significant difference in the VSS was observed between cimetidine-treated and untreated rats.
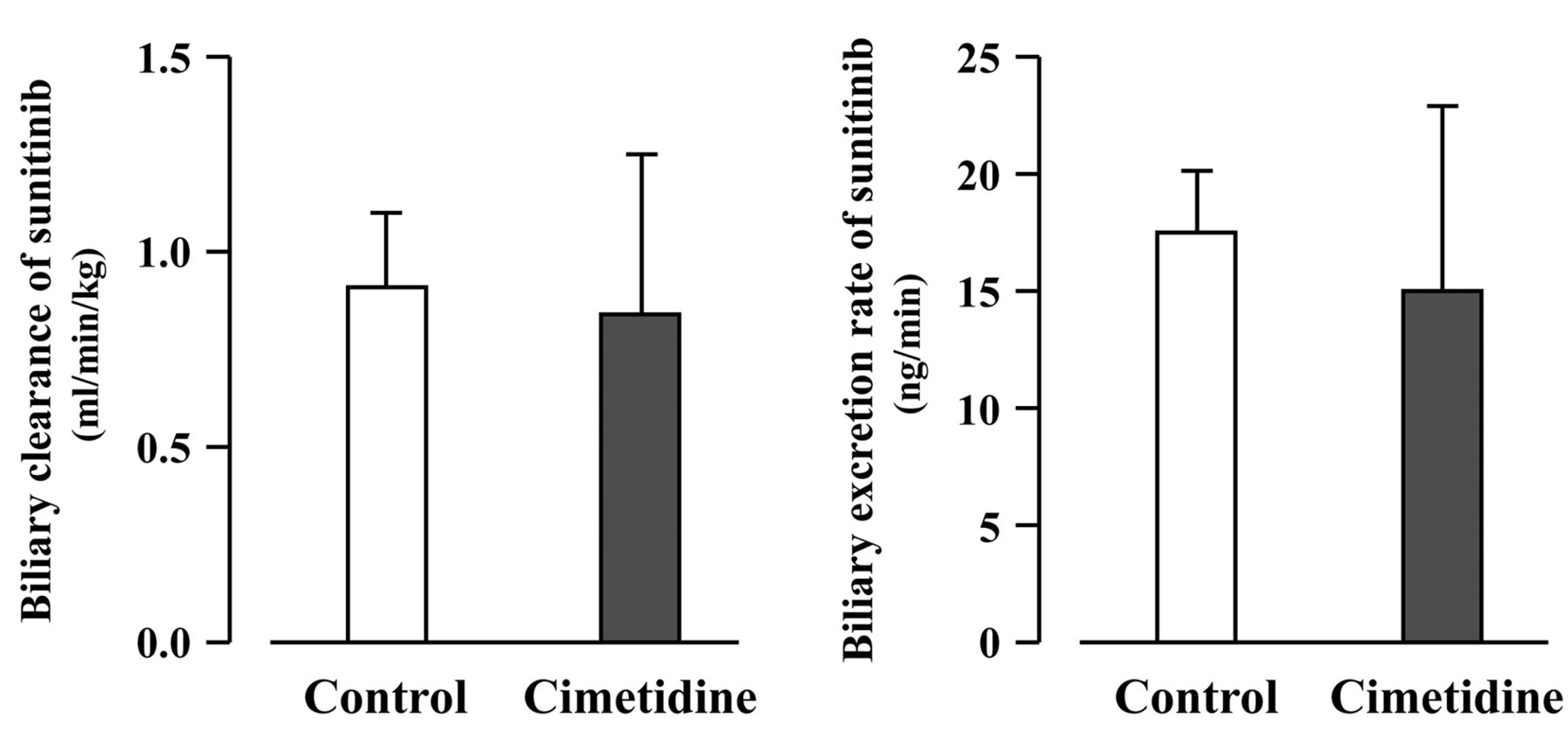
Effect of cimetidine on hepatobiliary excretion of sunitinib. The effect of cimetidine on biliary excretion of sunitinib under steady-state conditions in SD rats is illustrated in Figure 2. As shown in Figure 2, no significant differences in the CLBILE, biliary excretion rate (BER) and CSS were recorded between cimetidine-treated (0.84±0.41 ml/min/kg, 15.0±7.8 ng/min and 50.7±3.3 ng/ml, respectively) and untreated (0.91±0.19 ml/min/kg, 17.5±2.6 ng/min and 52.8±5.6 ng/ml, respectively) rats. The ratio of CLBILE to CLSYS of sunitinib was approximately 0.1.

Plasma concentration–time curves of sunitinib after a single intravenous administration (3 mg/kg) in cimetidine-treated and untreated Sprague-Dawley rats. Each point represents the mean±SD (n=3). Closed and open circles represent cimetidine-treated and untreated (control) rats, respectively. aSignificantly different from the control (p<0.05). When the standard deviation is small, it is included in the symbol.
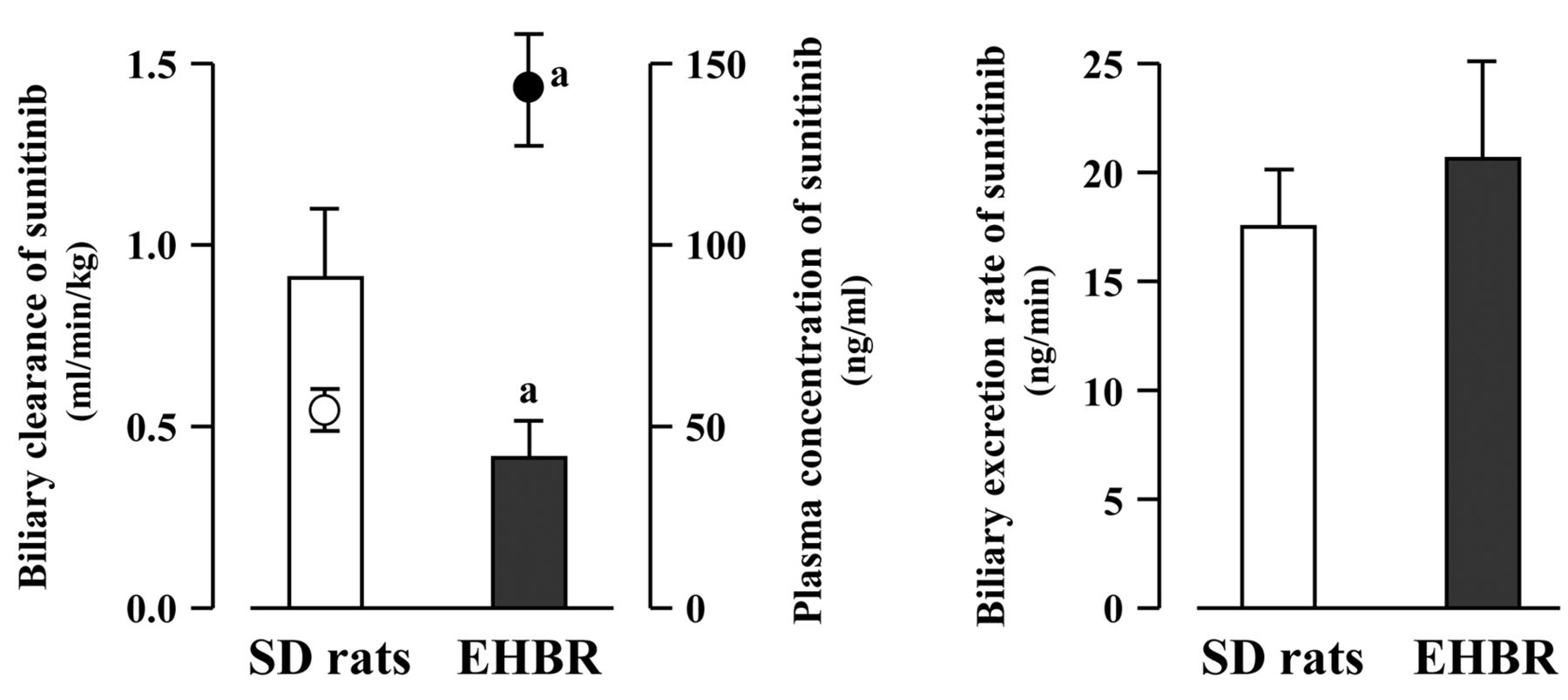
Contribution of Abcc2 to the biliary excretion of sunitinib in SD rats and EHBR. The contribution of Abcc2 to the biliary excretion of sunitinib was investigated using SD rats and EHBR (Figure 3). The bile flow rate in EHBR (12.7±3.5 μl/min) was significantly smaller than that in SD rats (24.7±2.8 μl/min). The CLBILE of sunitinib was significantly lower in EHBR (0.42±0.10 ml/min/kg) compared to SD rats (0.91±0.19 ml/min/kg). The CSS of sunitinib in EHBR (143.5±15.4 ng/ml) was significantly higher by 2.7-fold compared with that in SD rats (52.8±5.6 ng/ml). However, there was no significant difference in the BER of sunitinib between SD rats (17.5±2.6 ng/min) and EHBR (20.6±4.5 ng/min).
Discussion
Sunitinib, which is used for the treatment of renal cell carcinoma and gastrointestinal stromal tumor, commonly causes various side-effects, such as fatigue, nausea, diarrhea, anorexia, hypertension, and hand-foot syndrome. Uncommonly, a high plasma concentration of sunitinib leads to severe damage to the liver. Therefore, the dosage schedule of sunitinib and concomitant drugs, blood pressure and liver function should be routinely monitored because the incidence of such side-effects may be closely related to the pharmacokinetic behavior of sunitinib (4).
The present study focused on the effect of concomitant drugs on the pharmacokinetics of sunitinib. For example, concomitant use of sunitinib with ketoconazole, a potent CYP3A4 inhibitor, increased the area under plasma concentration–time curve of sunitinib by approximately 2-fold (20). On the other hand, the presence of rifampicin, a potent CYP3A4 inducer, dramatically reduces the plasma concentrations of sunitinib to approximately one-fourth (20, 21). There are no instructions on the pharmaceutical package insert regarding combination therapy with cimetidine, a typical CYP3A4 inhibitor, for patients receiving sunitinib therapy, although it is highly possible that drug interaction between sunitinib and cimetidine may occur. Therefore, we investigated the effect of cimetidine on the systemic elimination and steady-state hepatobiliary excretion of sunitinib in SD rats and EHBR.
Firstly, we investigated the effect of treatment with cimetidine on the plasma concentration–time curves of sunitinib after a single intravenous administration to SD rats. The results obtained showed that the treatment with cimetidine significantly delayed the terminal elimination of sunitinib with no change in the VSS. These findings suggest the possibility that the pharmacokinetics of sunitinib may be altered by co-administration of cimetidine. Therefore, the drug interaction between sunitinib and cimetidine may have been caused by inhibition of CYP3A4-mediated metabolism and/or efflux transporter-mediated biliary excretion of sunitinib.
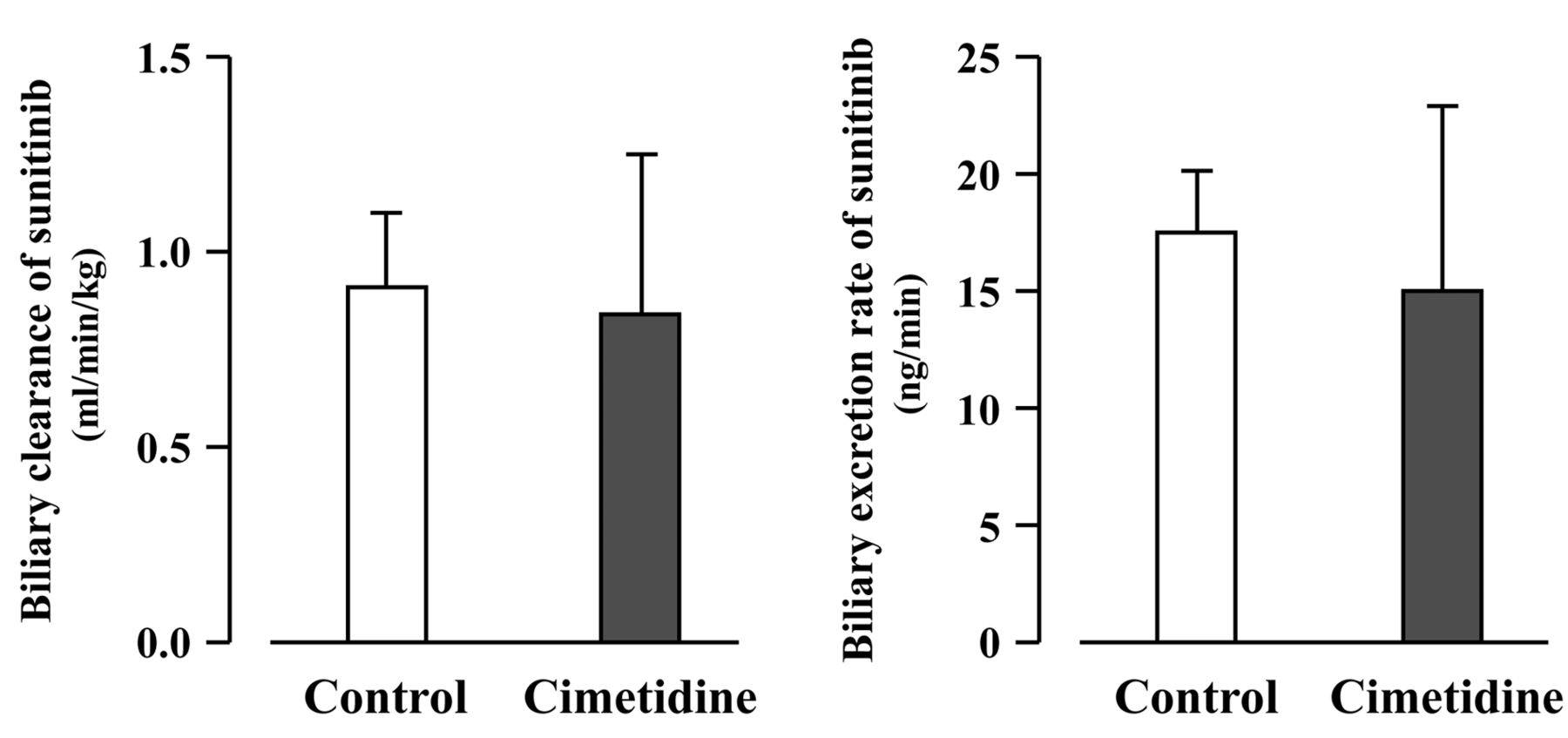
Effect of cimetidine on biliary clearance and biliary excretion rate of sunitinib in Sprague-Dawley rats. No significant difference was observed between cimetidine-treated and untreated rats. Each column represents the mean±SD (n=3).
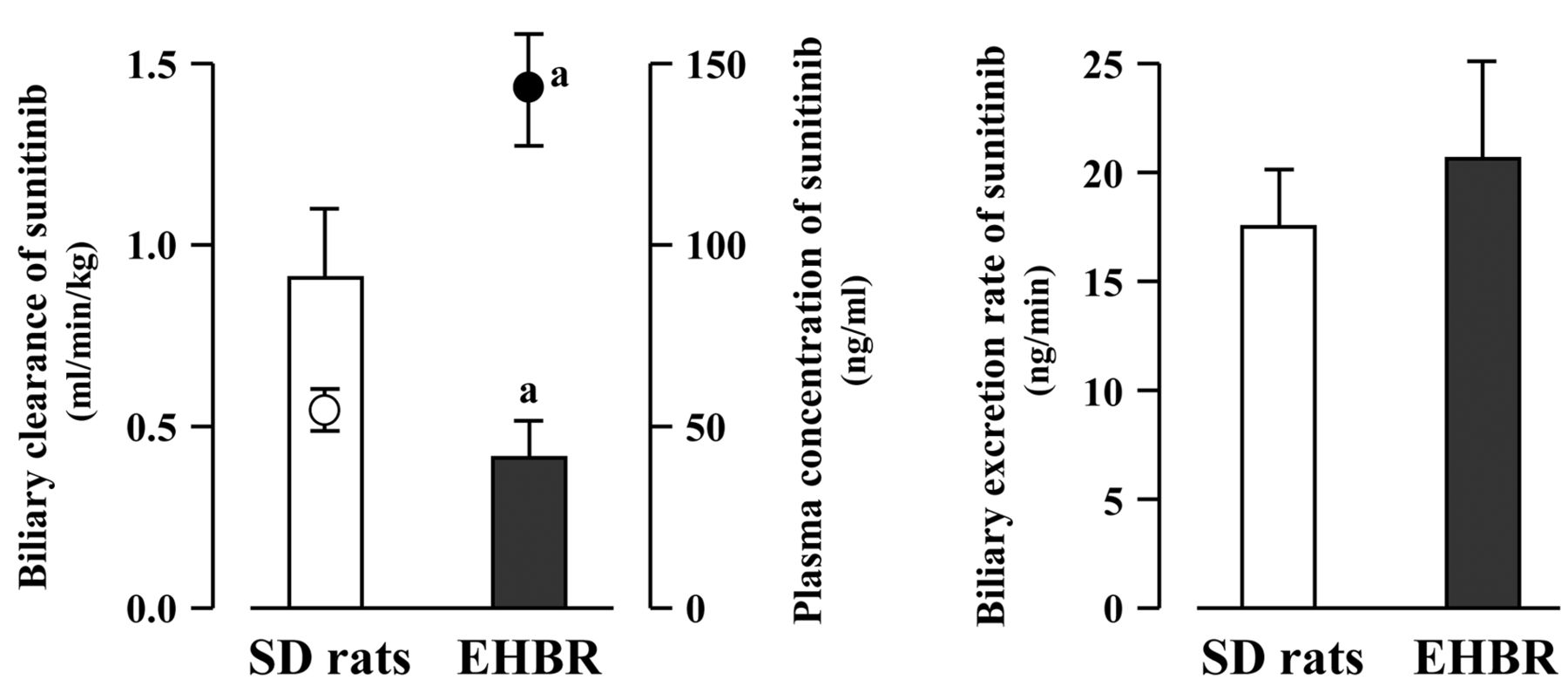
Biliary clearance, biliary excretion rate and steady-state plasma concentration of sunitinib in Sprague-Dawley (SD) rats and Eisai hyperbilirubinemic rats (EHBR). Each column represents the mean±SD (n=3). Closed and open circles represent plasma concentration of sunitinib in EHBR and SD rats, respectively. aSignificantly different from SD rats (p<0.05).
Secondly, we investigated the effect of cimetidine on the steady-state biliary excretion of sunitinib in SD rats with bile duct cannulation. In the biliary clearance experiments, it was demonstrated that cimetidine had no effect on the biliary clearance parameters, CLBILE and BER of sunitinib (Figure 2). These findings suggest that cimetidine has no effect on the biliary excretion of sunitinib by sunitinib-sensitive efflux transporters, including P-glycoprotein, although it is reported that cimetidine does inhibit P-glycoprotein. The ratio of CLBILE of sunitinib to CLSYS, which was calculated by dividing the maintenance dose of sunitinib by the steady-state plasma concentration, was approximately 0.1, suggesting that the contribution of biliary excretion of sunitinib to systemic elimination is small. It is likely that CYP3A4-mediated metabolism plays the major role in the systemic elimination of sunitinib.
Thirdly, we investigated the contribution of Abcc2 to the biliary excretion of sunitinib using SD rats and EHBR. The CLBILE of sunitinib in EHBR was dramatically and significantly lower than that in SD rats. However, there were no significant differences in the BER of sunitinib between SD rats and EHBR. These results suggest that sunitinib is not a substrate of Abcc2 and is instead excreted into the bile by other efflux transporters, such as P-glycoprotein and Abcg2. Considering that the CSS of sunitinib in EHBR was 2.7-fold higher than that in SD rats, it is likely that the decreased CLBILE observed in EHBR may have been caused by the increased CSS of sunitinib, although the precise reason for this cannot be explained at this stage.
There are some reports suggesting that Abcc3, an inducible efflux transporter, up-regulated in the basolateral membrane of hepatocytes in EHBR, plays a significant role in the basolateral export of Abcc3 substrates (22, 23). It is also reported that CYP enzyme activity, including Cyp3A2 (which corresponds to CYP3A4 in humans), is slightly but significantly lower in EHBR than SD rats (24). We, therefore, cannot exclude the possibility that the higher plasma concentration of sunitinib observed in EHBR may have been caused by the increased efflux of unchanged sunitinib from the hepatocytes into the blood by the presence of Abcc3 or an Abcc3-like sunitinib-sensitive protein in EHBR, so as to prevent the accumulation of sunitinib in the liver.
It is reported that tyrosine kinase inhibitors are substrates of various ABC transporters, including P-glycoprotein and ABCG2 (25-30). It is also reported that sunitinib is actively taken up from blood into hepatocytes by the organic anion transporting polypeptide isoforms expressed in the basolateral membranes of hepatocytes in the rat (31), Oatp1 and Oatp2, which correspond to human OATP1B1/SLCO1B1 and OATP1B3/SLCO1B3. It has been reported that hepatic protein expression of both Oatp1 and Oatp2 in EHBR is approximately one-half of that in SD rats (32). In the present study, the CLSYS of sunitinib in EHBR was significantly lower by approximately 60% compared with SD rats, which was consistent with the expression levels of Oatp1 and Oatp2. On the basis of these findings, we assume that the higher plasma concentrations and reduced CLSYS of sunitinib observed in EHBR may have been, in part, caused by the lower expression of the hepatic uptake transporters, Oatp1 and Oatp2. Therefore, it is also likely that the majority of CLSYS of sunitinib in EHBR is dependent on the hepatic uptake of sunitinib.
In conclusion, as far as we know, the present study is the first to demonstrate that co-administration of cimetidine alters the pharmacokinetics of sunitinib by CYP3A4 inhibition, that sunitinib is not a substrate of Abcc2, and that the contribution of biliary excretion of sunitinib is small. Although the data obtained from this study cannot be extrapolated directly to humans, the present findings may provide some useful information for patients with cancer who are scheduled to receive sunitinib therapy.
- Received June 9, 2013.
- Revision received June 24, 2013.
- Accepted June 25, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}