


Abstract
Background: Vasoactive intestinal peptide (VIP) receptors are overexpressed in a broad variety of tumours. For the detection of these tumours, novel chemically modified and shortened VIP derivatives were designed. Materials and Methods: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA)-derivatised VIP analogues were radiolabelled with 111In and in vitro and in vivo behaviour was evaluated using stability and internalisation assays, as well as an initial biodistribution study. Results: Radiolabelling of the VIP analogues resulted in high radiochemical yields, without need for further purification steps. Stability of the VIP derivatives was variable and cell uptake studies in VIP receptor-positive cell lines revealed that only a limited number of derivatives were internalised. In the tumour mouse model, no specific tumour targeting was shown. Conclusion: Since the tested VIP derivatives exhibited impaired in vitro and in vivo characteristics alternative modifications to increase their stability while retaining receptor affinity should be considered to enable the use of synthetic VIP analogues for tumour targeting.
The targeted delivery of radionuclides and therapeutic agents to pathological sites has important implications in clinical research for the detection, diagnosis and therapy of various cancer types. Specific in vivo targets for these purposes are peptide receptors which are overexpressed on malignant tissue and expressed less on normal tissue (1). In vivo targeting of such receptors can be established by using radiolabelled so-called regulatory peptides which are present in the gut, the endocrine system or the lymphatic tissue. In particular, solid-phase peptide synthesis and bio-conjugation techniques offer easy ways to prepare peptide-based targeting molecules, each one with specific properties. Such small peptides allow fast tissue penetration and binding to the receptor of interest but also rapid (mostly renal) excretion of the unbound peptide, reducing possible side-effects for the patient. Derivatives of regulatory peptides, e.g. the 111In-labelled somatostatin-analogue octreotide, are already well established in routine clinical nuclear medicine (2).
Vasoactive intestinal peptide (VIP), which was first characterised in porcine duodenum by Said and Mutt (3), and the potential for its use in targeted imaging approaches was first investigated by Virgolini et al. (4). VIP is a hydrophobic 28-amino acid peptide with three lysine (Lys15, Lys20, Lys21), two arginine (Arg12, Arg14) and two tyrosine residues (Tyr10, Tyr22), an essential histidine residue at the N-terminus and an amidated C-terminus (5). The secondary structure of VIP is mostly helical with a central α-helix and a random coil structure at the N- and C-termini (6). Together with the structurally similar pituitary adenylate cyclase-activating peptide (PACAP), it maintains a variety of biological activities in the human body. VIP acts as a vasodilator, stimulates the secretion of various hormones and has influence on growth and proliferation of normal as well as malignant cells (7). There are two types of VIP receptors, VPAC1 and VPAC2, which have a high affinity for both VIP and PACAP (8). They belong to the class II subfamily of G-protein-coupled receptors. The VPAC1 receptor is found on normal tissue such as peripheral blood cells, the gastrointestinal tract, liver, lungs as well as kidneys (9) whereas VPAC2 receptors are distributed throughout the central nervous system and in peripheral tissues such as skeletal and smooth muscles, heart, pancreas and stomach and also like VPAC1 in blood vessels, lungs and kidneys (10, 11). At pathological sites, such as neuroendocrine tumours, adenocarcinomas, melanomas, breast cancer, pancreatic carcinomas, non-small cell lung cancer (NSCLC) or neuroblastomas, the VPAC1 receptor type is mainly expressed (12). Exceptions are benign smooth muscle tumours, so-called leiomyomas, where mostly VPAC2 receptors have been detected. Using these G-protein-coupled VIP receptors as targets for imaging and therapeutic approaches is very attractive as they are expressed at high densities on more tumour types and at higher densities than the gold-standard somatostatin receptor (13). However, the overexpression of VIP receptors in normal tissue such as lungs, central nervous system, intestine or liver may affect the clinical usefulness of VIP derivatives (14).
In a number of clinical studies, various VIP derivatives radiolabelled with different radionuclides have been used for tumour imaging of a broad variety of cancer types (9, 15, 16). The two tyrosine residues at position 10 and 22 in VIP enable labelling with 123I using the direct labelling approach (17). 123I-VIP scintigraphy has been used to image endocrine tumours of the gastrointestinal tract, pancreatic cancer, colorectal cancer and liver cancer. However, the usage of 123I-VIP in clinical routine has been limited by the complex radiolabelling procedures and purification steps involved and the expensiveness and availability of 123I, which is a cyclotron-produced radionuclide. Indirect labelling approaches using bifunctional coupling agents allowing labelling with different radiometals has advantages over direct labelling approaches because they allow a more stable labelling with a so-called residualising label. After receptor-mediated internalisation into the target cell, the peptide undergoes lysosomal degradation and the radiometal chelator complex is trapped inside the cell (18). 99mTc-VIP derivatives have been used for the imaging of colorectal cancer and showed promising results in experimental animal models (19). Radiolabelling with positron-emitting radionuclides allowing highly sensitive positron-emission tomographic (PET) imaging was also investigated, showing promising results (20). Moody et al. labelled a VIP analogue with 18F to examine its ability to localise breast cancer (7). Thakur et al. (21) and Zhang et al. (22) used 64Cu-labelled VIP derivatives for PET imaging of breast and prostate cancer.
Nevertheless, VIP is a pharmacologically potent molecule and even low doses in the sub-microgram range produce toxic effects, including hypertension, diarrhoea and bronchospasm (23). Therefore efficient and often challenging purification of the radiolabelled product is crucial in order to reduce adverse pharmacological side-effects (14). Another disadvantage is the rapid in vivo degradation of native VIP by proteolytic digestion and endopeptidases, leading to an in vivo half-life of less than one min (24, 25). Common ways of overcoming such stability issues are the alteration of the peptide sequence of the natural peptide, an approach which has been investigated in this study, or to use carrier molecules, such as nanoparticles, which encapsulate the peptide and protect it from enzymatic degradation (26).
The aim of the present work was the synthesis and characterisation of new VIP analogues with specific chemical modifications in their peptide sequence allowing conjugation with 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) suitable for labelling with different trivalent radiometals. We investigated minor modifications in the peptide sequence in an attempt to improve the in vivo stability and especially designed a shortened VIP analogue. Since it is known that His1 plays a major role in the biologic activity of VIP (27), the chelator DOTA was positioned either at the N-terminus using an unnatural amino acid as linker, or to the ε-amino function of lysine at the C-terminus to study the influence of the positioning of DOTA on the receptor interaction. The improved in vitro stability of the modified derivatives was examined by incubation in a buffered salt solution, a solution containing a chelating agent to test the propensity to transchelation and fresh human serum to evaluate the biological stability against enzymatic degradation. The in vitro binding properties of the VIP derivatives were tested on VIP receptor-positive rat lung membranes using 125I-VIP as a competitor. Cell uptake studies were performed with Chinese hamster ovary cells stably transfected with human VIP receptors (VPAC1/VPAC2). In the initial biodistribution study performed with the shortened VIP analogues, 111In-DOTA-VIP-A7 and 111In-VIP-A7-DOTA, the tumour targeting properties in a PANC-1 mouse tumour model were investigated.
Materials and Methods
Unless stated otherwise, reagents were of analytical grade, obtained from Sigma-Aldrich (Vienna, Austria) or VWR International GmbH (Vienna, Austria) and used as supplied with no further purification. 111In chloride was bought either from PerkinElmer (Waltham, MA, USA) or Mallinckrodt Medical BV (Petten, the Netherlands) and 125I-VIP was purchased from GE Healthcare (Little Chalfont, UK).
Cell culture media, including Gibco® Ham's F12 Nutrient Mixture and Gibco Roswell Park Memorial Institute 1640 (RPMI 1640), Gibco fetal bovine serum, penicillin/streptomycin and penicillin/streptomycin/glutamine solutions (all Life Technologies, Carlsbad, CA, USA) were obtained from Thermo Fisher Scientific (Waltham, MA, USA).
Radioactivity measurements were carried out using the 2480 WIZARD2 Automatic Gamma Counter from PerkinElmer.
Synthesis of VIP derivatives. Based on natural VIP, seven different DOTA-conjugated VIP derivatives were synthesised. Modifications were the introduction of the oxidised form of methionine (Met) [M(O)] and the substitution of alanine with diphenylalanine (Dip). To minimise sterical influence on receptor binding, DOTA conjugation at the N-terminus was performed using the unnatural amino acid aminohexanoic acid (Ahx) as linker, whereas at the C-terminus, the side chain ε-amino function of an additional lysine residue was used. Besides introduction of the oxidised Met residue or a hydrophobic Dip in the proximity of Met, an N-terminally truncated peptide sequence was also investigated (VIP-A7). The detailed peptide sequences of the different VIP analogues are displayed in Table I.
Peptide sequences of the seven VIP analogues.
The synthesis of the various VIP derivatives was performed using a solid-phase peptide synthesis technique and a 9-fluorenylmethyloxycarbonyl (Fmoc)-strategy with a commercially available batch peptide synthesizer (PSSM-8; Shimadzu, Kyoto, Japan). In situ activation was carried out by using hydroxybenzotriazole/N,N’-diisopropylcarbodiimide. Briefly, during synthesis the growing peptide chain was covalently fixed on a polymeric carrier via its C-terminus. To obtain the C-terminal carboxamide after resin cleavage, an appropriate anchor group (peptide-amide linkage) was used at the polymeric support (TentaGel® S RAM resin; RAPP Polymer, Tuebingen, Germany). The temporary Fmoc protection group was cleaved with 30% piperidine in N,N-dimethylformamide. The side chain protection for Asn, Gln, His, Ser, Tyr, Thr, Asp, Arg and Lys was either trityl, tert-butyl, tert-butyloxycarbonyl (Boc) or 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Pbf). For derivatives with DOTA conjugated to the C-terminus using an additional lysine, the ε-amino function was protected with N-[1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl] (Dde). The primary amino function of the last amino acid in VIP-DOTA was protected with Boc. Reagents were used in a 5-fold excess and at high concentrations to drive reactions as close to completion as possible. VIP-M(O)-DOTA, VIP-A6-DOTA and VIP-A7-DOTA were acetylated at the N-terminus using a mixture of pyridine/acetic anhydride (10:1). For DOTA-VIP-M(O) and DOTA-VIP-A6, the unnatural amino acid Ahx acted as a linker at the N-terminus.
As already mentioned the position of DOTA was varied in the derivatives. Conjugation of DOTA after completion of the synthesis of the peptide sequence was carried out as follows.
Conjugation of the tert-butyl-protected DOTA ligand via the ε-amino function of Lys [VIP-DOTA, VIP-M(O)-DOTA, VIP-A6-DOTA, VIP-A7-DOTA]: The removal of the Dde protection group of Lys was performed using a mixture of 2% hydrazine hydrate solution in 1-methyl-2-pyrrolidinone. The progress was followed by a test cleavage [high pressure liquid chromatography (HPLC) and mass spectrometry (MS)]. After successful side chain deprotection, the tert-butyl-protected DOTA-ligand, 1,4,7,10-tetraazacyclododecane-1,4,7-tris(t-butyl acetate)-10-acetic acid, was coupled to the resin by help of benzotriazole-1-yl-oxy-tris-pyrrolidinophosphonium hexafluorophosphate (PyBOP), followed by another test cleavage to control completeness of the coupling.
Conjugation of the DOTA ligand via the N-terminus [DOTA-VIP-M(O), DOTA-VIP-A6, DOTA-VIP-A7]: The tert-butyl-protected DOTA was coupled to the resin-attached peptide via PyBOP activation. Completeness of synthesis was controlled by test cleavage.
Cleavage from the resin as well as side chain deprotection was performed in one step using a mixture of 94% trifluoroacetic acid with 2.5% water, 2.5% 1,2-ethanedithiol and 1% trifluoroisopropylsilane. After cleavage, the mixture was filtered via a sintered glass funnel and the filtrate was concentrated before the peptide was precipitated by adding diethyl ether to the cold solution. Repeated washing of the precipitate with cold diethyl ether, followed by dissolving the peptide in water and freeze-drying resulted in a white to off-white lyophilisate. Purity and identity of the crude product were determined by reversed-phase HPLC and MS. The purification of the crude peptide was carried out using a preparative reversed-phase HPLC system (LC-8A preparative liquid chromatograph; Shimadzu, Kyoto, Japan) with an appropriate gradient system containing water/acetonitrile mixtures using trifluoroacetic acid as ion pairing agent. All fractions which met the specifications for HPLC purity were pooled, solved in water and lyophilised to give the peptide as a trifluoroacetic acid salt.
Radiolabelling. For radiolabelling with 111In chloride at high radiochemical yield the DOTA-conjugated VIP derivatives (70 μg) were incubated with 35 to 50 μl 111In chloride solution (approximately 60 MBq in 0.05 M hydrogen chloride) and 50 μl buffer solution containing sodium acetate trihydrate (0.40 M) and gentisic acid (0.24 M) adjusted to pH 5.0. The labelling solution (total volume 150 μl) was then left to react for 25 min at 95°C. Radiochemical yield was assessed by reversed-phase HPLC on a Dionex P680 HPLC pump with a Dionex UVD 170 U UV/VIS detector (Dionex, Germering, Germany) and a Bioscan radiometric detector (Bioscan, Inc, Washington, D.C., USA). A Nucleosil® 120-5 C18 250×4.6 mm column (Macherey-Nagel GmbH & Co. KG, Dueren, Germany), with flow rates of 1.5 ml/min and UV detection at 220 nm were employed with the following gradient: acetonitrile (ACN)/0.1% trifluoroacetic acid/water: 0-15 min 0% ACN, 15-19 min 60% ACN, 19-22 min 0% ACN.
Cell culture. All cell lines were purchased from the European Collection of Cell Cultures (Salisbury, UK). For internalisation studies, Chinese hamster ovary cells stably transfected with human VPAC1 or VPAC2 receptors were cultured in tissue culture flasks (Cellstar®; Greiner Bio–One, Kremsmuenster, AUT) and Ham's F12 medium supplemented with 10% volume/volume (v/v) heat-inactivated fetal bovine serum and 1% (v/v) penicillin/streptomycin solution. Human pancreatic duct epithelioid carcinoma cells (PANC-1) were also grown in tissue culture flasks (Greiner Bio–One) and maintained in RPMI-1640 supplemented with 10% (v/v) heat-inactivated fetal bovine serum and 1% (v/v) penicillin/streptomycin/glutamine solution. Both cell lines were grown to confluence at 37°C in a humified atmosphere of 95% air/5% carbon dioxide. Culture media were replaced every two days.
In vitro characterisation. Distribution coefficient: Approximately 5 kBq of 111In-labelled VIP derivatives diluted in 0.5 ml phosphate dihydrate/1.4 mM monopotassium phosphate/136.9 mM sodium chloride phosphate-buffered saline (PBS; pH 7.4) were added to 0.50 ml octanol. The mixture was then vigorously vortexed for 15 min. Subsequently, triplicates of both the aqueous and octanol layer were collected in plastic vials. Radioactivity of the samples was measured in a gamma counter and the distribution coefficient was calculated.
Stability studies: The in vitro stability of 111In-labelled VIP analogues was tested by incubation in PBS, a PBS solution containing 4 mM diethylenetriaminepentaacetic acid (DTPA), and in fresh human serum at 37°C over a period of 24 h. The radioligands were incubated at a concentration of 1.5 μM and at preselected time points aliquots of PBS and DTPA solution were injected directly into the reversed-phase HPLC system to analyse the stability of the radiometal complex. The serum samples were precipitated with acetonitrile and centrifuged at 19,745× g for three min. Supernatant was taken off the vial and injected into the HPLC system to investigate the stability of the radiopeptides against enzymatic degradation.
Competitive binding studies: Three adult female Lewis rats of about 300 g (Charles River Laboratories, Sulzfeld, Germany) were used for the extraction/preparation of membrane fractions from pulmonary tissue known to express both VIP receptor subtypes (28). All procedures were performed according to the current international legislation and the Austrian animal protection law. Rats were anesthetised and sacrificed by intravenous injection of an overdose of the anaesthetic Thiopental (10 mg/kg BW). Lungs were excised and immediately put into ice-cold Hank's balanced salt solution containing 2 ml penicillin/streptomycin/neomycin solution and 0.2 ml aprotinin solution. Subsequently, lungs were homogenised using an Ultra-Turrax homogenizer (IKA Labortechnik, Staufen, Germany) followed by a centrifugation step at 500× g for 10 min at 4°C. Thereafter, the supernatant was removed and placed on ice. The remaining pellet was resuspended in 20 ml homogenisation buffer of 25 mM tris(hydroxymethyl) aminomethane buffer (pH 7.5) containing 0.3 M sucrose, 1 mM ethylene glycol tetraacetic acid, 0.25 mM phenylmethylsulfonyl fluoride and 0.5 ml aprotinin solution. The homogenate was centrifuged again at 500× g for 10 min at 4°C. This procedure was repeated three times. The combined supernatant was then centrifuged at 48,000× g for 45 min at 4°C. The supernatant was discarded and the pellet was washed twice with washing buffer of 50 mM tris(hydroxymethyl)aminomethane buffer (pH 7.5) containing 5 mM magnesium chloride, 0.01 mM bacitracin solution, 25 mM phenylmethylsulfonyl fluoride and 2.5 ml aprotinin solution. The final pellet was resuspended in 1 to 2 ml washing buffer and protein concentration of the sample was determined using a spectrometric method (Bradford assay). The membranes were distributed into tubes, each aliquot containing enough protein for one binding assay.
The in vitro receptor binding affinity of native VIP (control) and the seven VIP analogues was determined in competition with 125I-VIP. One day before the experiment, 96-well Nunc® multiscreen plates (Thermo Fisher Scientific) were coated with 1% polyethylenimine in water for 2-3 h at room temperature to reduce nonspecific binding of radioligand to the wells. Each well was then washed twice with water and the multiscreen plates were put in an oven (37°C) and left to dry overnight. Thereafter, wells were washed with 250 μl ice-cold dilution buffer of 20 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid buffer (pH 7.3) containing 1% bovine serum albumin, 40 μl of 10 M magnesium chloride solution and 40 μl of 14 mM bacitracin-solution. Then 50 μl of different concentrations of native VIP or VIP analogues (0.01 to 1000 nM concentration in the assay) and a fixed concentration of the competing radiolabelled standard (125I-VIP; 30,000 cpm) were put into each well. As a last step 100 μl of lung membrane (40 to 50 μg membrane/well) were added to the wells and the plates were incubated on a shaker for 1 h at room temperature. The reaction was interrupted by rapid filtration of the solution through the filters and rinsing with ice-cold 10 mM tris(hydroxymethyl)aminomethane buffer. Filters were removed and accumulated radioactivity measured in a gamma counter. The half-maximal inhibitory concentration (IC50) values were calculated by fitting the non-linear regression using the Origin Software (OriginLab Corporation, Northampton, MA, USA).
Internalisation studies: For determination of the receptor-mediated radioligand uptake, VPAC1 and VPAC2 cells were counted to a density of 1.5×106 cells, seeded into 6-well plates (Greiner Bio-One) and left to grow for 48 h. On the day of the experiment, the medium was removed and cells were washed twice with 1 ml ice-cold Ham's F12 medium containing 1% fetal bovine serum. After the addition of 1.2 ml medium, either 150 μl of PBS/0.5% bovine serum albumin solution (total series) or 150 μl of 10 μM VIP (blocking solution; nonspecific series) in PBS/0.5% bovine serum albumin solution was added to each well. Then the corresponding 111In-labelled VIP derivatives (approximately 30,000 cpm; 1 nM) in PBS/0.5% bovine serum albumin solution were added and the plates incubated for 30, 60 or 120 min at 37°C. Incubation was interrupted by removal of medium and cells were washed twice with 1 ml ice-cold Ham's F12 medium and 1 ml 0.05 M glycine buffer (pH 2.8) was put into each well. After five min incubation, the supernatant (membrane-bound radioligand) was collected in plastic vials and the procedure was repeated. Cells were then lysed with 1 ml of 1 N sodium hydroxide solution. Internalised (sodium hydroxide) and membrane-bound (glycine) fractions were determined by measuring radioactivity in a gamma counter. Protein content in the sodium hydroxide fraction was calculated using a spectrometric method (Bradford assay). The internalised activity was expressed as percentage of the total activity per milligram protein.
In vivo characterisation. Biodistribution studies: All conducted animal experiments were performed in accordance with the regulations of the Austrian animal protection law and with the permission of the Austrian Ministry of Science (BMWF-66.011/0147-II/10b/2008).
For initial biodistribution studies 6-week-old, female athymic BALB/c nude mice (Charles River Laboratories) were used. Mice were maintained under pathogen-free conditions on a normal ad libitum diet. For induction of the tumour xenografts, 5×106 PANC-1 cells were injected subcutaneously into the right hind limb of the mouse. The state of health of the animals and tumour size was checked on a regular basis. The biodistribution studies were performed after two to four weeks when tumours had reached a size of 0.5 to 1 mm3. Into a lateral tail vein mice were injected with 0.1 MBq (5 MBq 111In/kg bodyweight; 0.1 μg peptide/mouse) of 111In-DOTA-VIP-A7 or 111In-VIP-A7-DOTA, respectively. During the incubation time, animals were kept warm to avoid hypothermia. At 1 or 4 post injection (p.i.) animals were sacrificed by cervical dislocation. Subsequently, tissue (blood, muscle) and organs (heart, lungs, liver, spleen, pancreas, stomach, intestine, kidneys) were removed and transferred to preweighed plastic tubes and weighed. The content of the stomach was removed and the emptied organ weighed. Standards of the injected radioligand were prepared and the radioactivity of the various probes and the standards was measured in a gamma counter and the percentage of injected dose per gram tissue/organ (% ID/g) was calculated.
Results
Peptide synthesis and radiolabelling. The different DOTA-conjugated peptide derivatives were obtained in 20 to 30% yield. A chemical purity ≥95% was confirmed by reversed-phase HPLC and matrix-assisted laser desorption/ionization (MALDI) time-of-flight (TOF) MS.
It was possible to label all derivatives with 111In chloride at high radiochemical yields (>95%) and no further purification steps were necessary. The following radiochemical yield values were obtained: 98.1±1.6% for VIP-DOTA (n=4), 97.1±1.0% for DOTA-VIP-M(O) (n=5), 99.2±1.2% for VIP-M(O)-DOTA (n=3), 98.2±1.7% for DOTA-VIP-A6 (n=3), 99.8±0.3 for VIP-A6-DOTA (n=4), 99.7±0.6% for DOTA-VIP-A7 (n=4) and 99.7±0.5% for VIP-A7-DOTA (n=5).
In vitro characterisation of the radioligands. The distribution coefficients of the VIP analogues were as follows: -3.1 for VIP-DOTA, -2.5 for DOTA-VIP-M(O), -2.7 for VIP-M(O)-DOTA, -2.6 for DOTA-VIP-A6, -2.8 for VIP-A6-DOTA, -3.4 for DOTA-VIP-A7 and -2.9 for VIP-A7-DOTA indicating a high hydrophilicity for all derivatives.

In vitro stability testing of the seven 111In-labelled VIP derivatives in fresh human serum (1.5 μM peptide/1 ml serum) at selected time points. Values are expressed as the percentage of intact radioligand [Dip: diphenylalanine; DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; M(O): oxidised form of methionine; Met: methionine; VIP: vasoactive intestinal peptide].
The in vitro stability studies in PBS, fresh human serum and against trans-chelation (DTPA solution) showed variable values for the different radiolabelled VIP analogues. In fresh human serum, the Met17-oxidised derivatives, namely 111In-DOTA-VIP-M(O) and 111In-VIP-M(O)-DOTA, had the highest stability, whereas the lowest stability was found for the shortened derivatives 111In-DOTA-VIP-A7 and 111In-VIP-A7-DOTA (Figure 1). After 30 min, the percentage of intact peptide had decreased to values <90% and after 24 h, for all VIP analogues, less than 7% of intact peptide was detected. In PBS and DTPA solution the various VIP derivatives exhibited higher stabilities. All 111In-labelled VIP analogues were found at >75% 120 min post preparation, with the exception of 111In-DOTA-VIP-A7 (<70% of intact peptide). After 24 h incubation, values dropped to around 50 to 80% of intact peptide (Table II). All in all, no relevant differences in stability due to the position of DOTA at the C- or N-terminus, or dependent on the various applied modifications of the peptide sequence were observed.
Receptor binding and cell uptake studies. The in vitro receptor binding assay on rat lung membranes using increasing amounts of native VIP and VIP derivatives in competition with 125I-VIP revealed variable IC50 values. In comparison with native VIP, which was used as control, with an IC50 value of 2.8 nM confirming high receptor affinity, only for the full-length VIP analogues with C-terminal DOTA-conjugation was binding affinity maintained. A high binding affinity was found only for VIP-DOTA with an IC50 value of 5.6 nM. VIP-A6-DOTA, with Dip in position 18, exhibited a medium receptor binding (IC50 value <50 nM). A low binding affinity was also found for VIP-M(O)-DOTA with Met(O) in position 17 (IC50 value <100 nM). For all other derivatives with an N-terminal DOTA-conjugation [DOTA-VIP-M(O), DOTA-VIP-A6], as well as with the shortened peptide derivatives (DOTA-VIP-A7, VIP-A7-DOTA) the receptor binding characteristics were found to be strongly impaired by the structural changes (IC50 values >1000 nM).

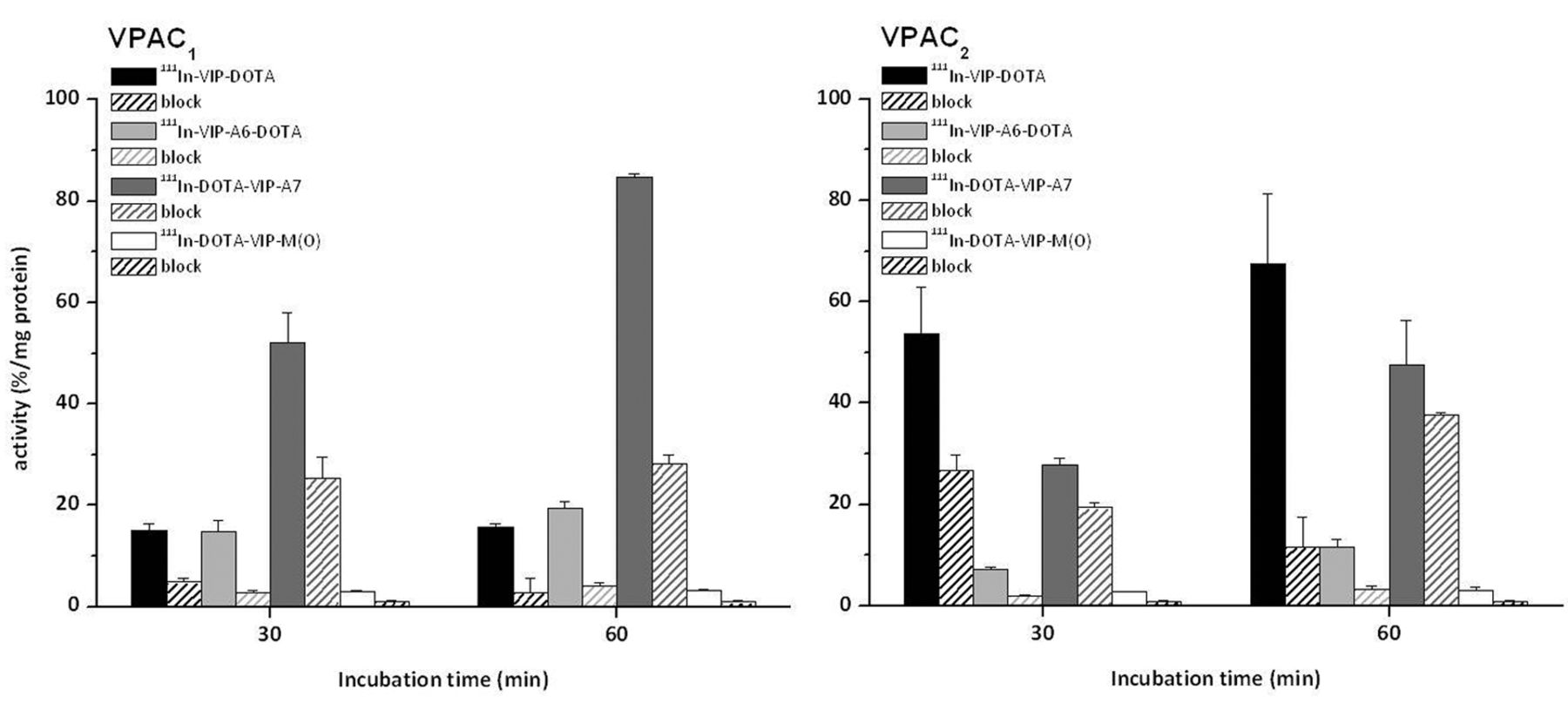
Cell uptake studies of different 111In-labelled VIP analogues on VPAC1 and VPAC2 receptor-positive cells after 30 and 60 min incubation: 111In-VIP-DOTA and 111In-DOTA-VIP-A7 had a much higher cell uptake in comparison with 111In-DOTA-VIP-M(O) and 111In-VIP-A6-DOTA, with an impaired receptor affinity. For the blocking studies 10 μM native VIP was used. Values are expressed as means±standard deviation (n=5). [DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; M(O): oxidised form of methionine; VIP: vasoactive intestinal peptide].
In vitro characteristics of different 111In-labelled VIP analogues. Values are expressed as the percentage of intact peptide.
The cell uptake studies using VPAC1 and VPAC2 receptor-positive cells showed variable results for the different VIP analogues (Figure 2). Interestingly, in VPAC1 cells, the highest uptake was found for 111In-DOTA-VIP-A7 (52.2±4.8 and 84.6±0.8% of total activity/mg protein at 30 and 60 min after incubation) which showed a reduced receptor affinity in rat lung membranes. For 111In-VIP-DOTA (15.8±0.6% at 60 min) and 111In-VIP-A6-DOTA (19.5±1.2% at 60 min), a high cell uptake was also shown. Blocking studies with natural VIP resulted in a reduction of the uptake of 111In-VIP-DOTA and 111In-VIP-A6-DOTA by 67 to 82%, showing that this uptake was receptor specific. For 111In-DOTA-VIP-A7, a lower reduction of the uptake (by 51 to 67%) was obtained by co-incubation with natural VIP. 111In-DOTA-VIP-M(O) had a minor cell uptake of 3.3±0.2% at 60 min which was blocked by 71% by natural VIP. The cell internalisation into VPAC2 cells was the highest for 111In-VIP-DOTA (53.7±9.3% and 67.6±13.8% at 30 and 60 min after incubation) and 111In-DOTA-VIP-A7 (27.8±1.3% and 47.7±8.7% at 30 and 60 min after incubation). In blocking studies, the uptake of 111In-VIP-DOTA was reduced by up to 83%, whereas the uptake of 111In-DOTA-VIP-A7 could not be reduced by more than 30%, indicating a highly nonspecific binding. 111In-VIP-A6-DOTA led to a cell uptake of 11.5±1.6% after 60 min incubation which was blocked by up to 73% by co-incubation with natural VIP. Similarly as observed in VPAC1 cells, 111In-DOTA-VIP-M(O) led to a minor cell uptake of 3.1±0.6% at 60 min which was again blocked by 71% by natural VIP. For 111In-labelled DOTA-VIP-A6, VIP-A7-DOTA and VIP-M(O)-DOTA, no specific or very low uptake (<1.5% of total activity/mg protein) and no differences between the two cell lines were found (data not shown).
In vivo characterisation. Biodistribution studies: To study the in vivo properties of the shortened VIP derivatives, an initial biodistribution study of 111In-DOTA-VIP-A7 and 111In-VIP-A7-DOTA using BALB/c nude mice bearing PANC-1 tumour xenografts was carried out (Table III).
For 111In-DOTA-VIP-A7 and 111In-VIP-A7-DOTA the major uptake of radioactivity at 1 hour p.i. was found in the kidneys. These values increased only slightly for both VIP-A7 derivatives at 4 hours p.i.. For 111In-VIP-A7-DOTA the uptake in the liver was also high whereas 111In-DOTA-VIP-A7 showed lower liver uptake. The amount of accumulated activity in the spleen was found to be two times higher for 111In-VIP-A7-DOTA at 1 h p.i. than for 111In-DOTA-VIP-A7 and even three times higher at 4 h p.i.. Blood levels of both VIP derivatives were low at approximately 0.3% ID/g. The nonspecific uptake in the other tissue/organs was comparable and was for all samples <3% ID/g. The uptake in the PANC-1 tumour xenograft was found to be strongly impaired, with values below 1.5% ID/g for both 111In-DOTA-VIP-A7 and 111In-VIP-A7-DOTA.
Discussion
Targeting cell surface receptor-expressing tumour cells with radiolabelled peptides is a well-established method in nuclear medicine especially for the diagnosis and therapy of different tumour types. So far VIP has been used for a variety of clinical applications including the diagnosis of various tumours or neurodegenerative diseases (29).
Biodistribution in PANC-1 tumour-bearing BALB/c nude mice of 111In-labelled DOTA-VIP-A7 and VIP-A7-DOTA 1 and 4 h after injection. Values are expressed as the percentage of injected dose per gram (means±standard deviation; n=12).
Since VIP specifically binds mainly to VPAC1 receptors expressed at high densities on many tumour types, the aim of this study was to develop a novel radiolabelled VIP analogue derivatised with DOTA for radiometal-labelling with improved stability against enzymatic degradation in vivo and high specific binding to the VPAC1 receptor. The DOTA-derivatised VIP analogues were radiolabelled with 111In with high radiochemical yields (>97%) without need for further purification steps. The straightforward labelling procedure with 111In presents a clear advantage compared to the complex labelling procedure of 123I-VIP (17).
For the design of new VIP analogues using chemical modifications such as truncation of the peptide sequence or substitution of individual amino acids with unnatural amino acids, it is also crucial to know the mechanism of the peptide binding to its corresponding receptor and how the sequential arrangement of amino acids along the peptide sequence influences the biological behaviour of the derivatives (30). Although the exact mechanism of VIP binding to its receptor is not yet known various authors claim that for binding of VIP to the VPAC1 receptor, three residues (Arg14, Lys15, Lys21) interact with the acidic receptor residues (Glu36, Asp68, Asp196) through electrostatic interactions (29, 31). As already mentioned, His1, as well as Phe6, of VIP play a general role in the biological activity of the peptide (32). Based on this knowledge, these residues were not changed within the sequence of the VIP derivatives under investigation. Two minor chemical modifications were attempted in the peptide structure of native VIP with the aim of improving the problems of in vivo stability caused by rapid proteolytic degradation. VIP analogues with modified amino acid sequence, resulting in a more stable peptide conjugate, which could possibly be used as a therapeutic agent for bronchial asthma or for improved in vivo imaging of VIP-expressing tumours, have been reported by other groups and indicate the possibility of chemical modifications in position 17 and 18 (33-35). In the present study, oxidation of Met in position 17 in VIP-M(O) and substitution with Dip in position 18 in VIP-A6, however, actually reduced the receptor binding on rat lung membranes. Only when combined with C-terminal DOTA conjugation were peptide derivatives with reasonable binding affinity (IC50 value <50 to 100 nM) obtained. The abolishment of receptor binding for N-terminal DOTA-conjugation is in accordance with previous studies of Dangoor et al. (6), who showed that glycosylation of VIP near the N-terminal region (Ser2, Thr7 or Thr11) resulted in a considerable loss of binding affinity. The random coil structure at the N-terminus of VIP was demonstrated as being crucial for biological activity and specific receptor recognition (36).
For both the modifications applied in the present study, only minor chemical stabilisation against enzymatic degradation was found. Met oxidation seemed to be superior in stabilising the peptide analogue, whereas substitution with Dip had no effect on stability but also had a lower effect on receptor binding. The oxidation of Met in position 17 was reported to reduce the biological activity of VIP due to the disruption of the long α-helical structure of VIP (37). For these reasons, in the shortened VIP derivative VIP-A7, substitution with Dip in position 18 was chosen as the chemical modification. However, the receptor binding of both DOTA-VIP-A7 and VIP-A7-DOTA was strongly reduced (IC50 value >1000 nM). This is in contrast to earlier studies reported by Tams et al., showing a more than 300-fold improvement in binding affinity for truncated VIP derivatives with Dip in position 18 (38).
In the cell uptake studies using VPAC1 and VPAC2 receptor-positive cells, only with C-terminal DOTA-conjugated 111In-VIP-DOTA and 111In-VIP-A6-DOTA was receptor-specific cell uptake observed in both cell lines, whereas 111In-VIP-M(O)-DOTA and 111In-VIP-A7-DOTA were not internalised. The low specific cell uptake of 111In-DOTA-VIP-M(O) and the high uptake of 111In-DOTA-VIP-A7, especially in VPAC1 cells (>80% of total activity/mg protein at 60 min after incubation), was an unexpected finding given the strongly impaired receptor affinity of all N-terminally DOTA-conjugated VIP derivatives. This high cell uptake seems rather to be related to a high nonspecific binding of 111In-DOTA-VIP-A7 and could possibly explain why no displacement of 125I-VIP was observed for DOTA-VIP-A7 and VIP-A7-DOTA in the displacement assays on rat lung membranes.
In the biodistribution study performed in BALB/c nude mice bearing PANC-1 tumour xenografts, 111In-DOTA-VIP-A7 and 111In-VIP-A7-DOTA exhibited fast blood clearance. The main uptake was found in kidneys, with values of up to >20-25% ID/g. The uptake in spleen and liver of 111In-DOTA-VIP-A7 was in the order of 2-8% ID/g and more pronounced for 111In-VIP-A7-DOTA (5-19% ID/g). The uptake values at 1 h p.i. in liver and kidneys were much higher than the uptake observed by Cheng et al. after administration of a full-length 18F-labelled [R8,15,21, L17]-VIP analogue in nude mice bearing colorectal tumours (30). Although Cheng et al. also found the highest amount of accumulated activity to be in the kidneys, the uptake in the spleen was <0.5% ID/g and in the liver <1% ID/g. In contrast to this, Zhang et al. reported the highest uptake activity in liver (>55% ID/g) and lungs (>34% ID/g) at 4 h p.i. with a full-length 64Cu-labelled VIP derivative (22). The liver uptake of our tested VIP derivatives was only half as high and the lung uptake was also much lower (<2% ID/g). Reasons for the varying distribution patterns are not yet known but might be related to the different radionuclides used in the studies. The uptake in the PANC-1 tumour xenografts of both VIP-A7 derivatives was very low (<1.5% ID/g), which seems to be related to a combination of impaired receptor affinity and low stability against enzymatic degradation in vivo. The lack of in vivo uptake cannot be explained by a possible antagonistic activity of the shortened VIP derivative used in this study (38) because no clear specific binding was shown on VPAC1 receptor-expressing rat lung membranes. These results indicate that the full-length VIP sequence is required for high in vivo tumour targeting (22, 30).
Conclusion
In this study, seven VIP analogues were synthesised and their stability and receptor binding affinity were characterised. The VIP analogues were labelled with 111In to investigate the influence of different chemical modifications of the peptide sequence on stability against enzymatic degradation, receptor affinity, and cell uptake, as well as tumour targeting. Although all VIP derivatives were efficiently labelled with 111In resulting in high radiochemical yields, their in vitro performance was only slightly improved compared to natural VIP. Only C-terminal DOTA-conjugation seemed to provide derivatives with high binding affinity. Despite the unexpected finding of high cell uptake of 111In-DOTA-VIP-A7, an initial in vivo biodistribution study with 111In-DOTA-VIP-A7 and 111In-VIP-A7-DOTA revealed no specific tumour targeting. Overall these data show the substantial challenges in the design of new VIP derivatives and indicate that in the development of alternative VIP derivatives for various applications in nuclear medicine, the chemical group used for radiolabelling should be preferentially conjugated at the C-terminus, whereas chemical modifications and truncation of the amino acid sequence with the aim of increasing the stability against enzymatic degradation and potentially also limit the high side effects in vivo need to be studied in more detail.
Acknowledgements
The Authors report no conflict of interest and are alone responsible for the content and writing of this article.
The Austrian Nano-Initiative co-financed this work as part of the Nano-Health project (number 0200), the subprojects NANO-NUC were financed by the Austrian FWF (Fonds zur Foerderung der Wissenschaftlichen Forschung; number N208-NAN) and NANO-LIPO was financed by the Austrian Research Promotion Agency FFG (project Nano-Health; number 819721).
- Received February 19, 2013.
- Revision received March 19, 2013.
- Accepted March 19, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.