


Abstract
Aim: Sorafenib is a promising treatment for hepatocellular carcinoma (HCC) but recent toxicity concerns suggest that new strategies for its use are needed. One approach for reducing toxicity is to use lower doses of sorafenib in combination with other complementary mechanisms. Celecoxib, a cyclooxygenase-2 inhibitor, has been shown to inhibit HCC, and we hypothesized that low-dose sorafenib co-administered with celecoxib could act synergistically in the inhibition of HCC. In this study, the effect of sorafenib was tested in combination with celecoxib on the growth of human HCC cells in vitro. Materials and Methods: Two human HCC cell lines, HepG2 and Huh7, were treated with sorafenib and celecoxib, alone and in combination, and the effect of these treatments on growth, apoptosis, and expression of phospho-AKT was evaluated by WST-8, DNA content, and immunocytochemical assays, respectively. Results: When compared to the actions of either agent alone, the combination of low concentrations of sorafenib (<5 μM) and celecoxib (<20 μM) resulted in enhanced inhibition of both cell growth and AKT activation, and increased the induction of apoptosis. Combination index (CI) analysis showed that the growth inhibition effect was synergistic. Conclusion: This study shows that celecoxib synergistically potentiates the sorafenib-mediated antitumor effect. This finding establishes the foundation for clinical trials evaluating the efficacy of co-administration of soerafenib and celecoxib as a treatment for HCC.
Hepatocellular carcinoma (HCC) is a major health problem, with over 700,000 new cases diagnosed each year worldwide (1). Globally, HCC is one of the most prevalent malignancies, accounting for approximately 6% of all human cancers and one million deaths annually (1, 2). The incidence of HCC in the United States has increased significantly in recent years (3). HCC patients with a surgically-resectable localized tumor have a much better prognosis, although there is still only a 20-51% 5-year survival rate (4). Clearly, there is an urgent need for new therapies for this aggressive disease.
Sorafenib is a small molecule that inhibits tumor-cell proliferation and tumor angiogenesis and increases the rate of apoptosis in a wide range of tumor models including HCC (5, 6). It acts by inhibiting the serine-threonine kinases, RAF-1 and B-RAF, and the receptor threonine kinase activity of the vascular endothelial growth factor receptors (VEGFRs) 1, 2, and 3 and the platelet-derived growth factor receptor (PDGFR-B). Cellular signaling mediated by the RAF-1 and VEGFR pathways has been implicated in the molecular pathogenesis of HCC, providing a rationale for investigating sorafenib for this indication (6). In pre-clinical experiments, sorafenib displayed antiproliferative activity in liver cancer cell lines, and reduced tumor angiogenesis and increased tumor cell apoptosis in a mouse xenograft model of human HCC (6). In a multicenter, phase-3, double-blind, placebo-controlled trial, sorafenib was demonstrated to be effective for advanced HCC (7).
One limitation of sorafenib use is its toxicity. A variety of adverse reactions to sorafenib have been reported, including diarrhea, hypertension, and nausea (8, 9). Most notably, cutaneous toxic effects include mucositis (20%), rash (19-40%), alopecia (27%), xerosis (16%), xerostomia (11%), and hand-foot skin reaction (HFSR) (20-30%) (11). The most clinically-significant and dose-limiting dermatological toxicity is HFSR, which may be associated with significant tenderness, affecting the function and quality of life, and may force the modification or discontinuation of critical anti-neoplastic therapy (10). In Japan, HFSR was apparent in 51% of cases (7% were grade 3) (11). One approach to overcome this toxicity is to use lower doses of sorafenib in combination with other complementary agents. However, success requires the identification of agents that can potentiate the sorafenib-mediated tumor inhibition without significant systemic toxicity. Such potentiation could also have cost-benefit advantages due to the high cost of sorafenib when given at therapeutic doses (12).
Non-steroidal anti-inflammatory drugs (NSAIDs) appear to reduce the risk of developing cancer (13). One mechanism through which NSAIDs act to reduce carcinogenesis is to inhibit the activity of cyclooxygenase-2 (COX-2), an enzyme that is overexpressed in various cancer tissues (14, 15). Recently, selective COX-2 inhibitors have been shown to act additionally through COX-2-independent mechanisms (16). Several reports have demonstrated that combination of celecoxib with other agents has synergistic anti-proliferative and pro-apoptotic effects in various types of cancers (17, 18).
Although anti-HCC activity has been shown for both sorafenib and celecoxib, the effect on HCC of these drugs in combination has not, to our knowledge, been reported. We hypothesized that these drugs may synergize to be more effective than either agents administered alone. In the present study, we tested whether co-administration of low-dose sorafenib with celecoxib could potentiate the inhibition of cell viability and, if so, through which mechanism.
Materials and Methods
Agents and reagents. Celecoxib and sorafenib were purchased from LC laboratories (Woburn, MA, USA). For immunofluorescence analysis, we used anti-phospho-AKT473 antibody (Cell Signaling Technology, Beverly, MA, USA).
Culture of tumor cells and viability assay. HepG2 and Huh7 cells were obtained from RIKEN Cell Bank (Osaka, Japan) and cultured in RPMI medium supplemented with 5% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin/streptomycin (Meiji Seika, Tokyo, Japan). Viable cells were counted using the Cell Counting Kit-8 (Dojindo, Osaka, Japan). Treatments were carried out with different concentrations of sorafenib, celecoxib, or dimethyl sulfoxide (DMSO) control. DMSO concentrations were <0.1%. For WST-8 proliferation assays, cells were cultured in a 96-well dish (NUNC) in a volume of 100 μl and a cell density of 5×104 cells/ml. Assays were run for 48 h using a minimum of five replicates. For co-incubation with drugs, cells were treated with different concentrations of sorafenib (0.01-100 μmol/l) and celecoxib (0.01-100 μmol/l). Proliferation assays were performed using the WST-8 cell proliferation kit (Dojindo) according to the manufacturer's instructions.
DNA content and flow cytometry. Cells were harvested after treatment by trypsinization, fixed with ice-cold 70% ethanol and incubated at 4°C for 1 h. Cells were then washed with phosphate buffered saline (PBS) and incubated with 25 μg/ml RNase for 30 min at room temperature, then incubated with 25 μg/ml propidium iodide (PI). Cell debris were excluded based on forward vs. side scatter. Data from 10,000 events were collected in the final gated histograms. Apoptotic cells were identified based on the hypodiploid DNA content (sub-G1 fraction) that results from DNA fragmentation. Cells were subjected to cell-cycle analysis by flow cytometry using an FC500 flow cytometer (Beckman Coulter, Paris, France).
Immunofluorescence microscopy. Cultured cells were seeded onto glass coverslips and allowed to adhere for 24 h under routine culture conditions. After this growth period, cells were incubated with sorafenib, celecoxib, or both. Cells were then formalin-fixed, incubated with primary then secondary antibodies, and a series of images were captured using an inverted microscope (I×81; Olympus, Tokyo, Japan). Fluorescence analyses were performed using Lumina Vision (Mitani Corporation, Fukui, Japan).
For analysis of phospho-AKT expression in HCC cells, a phospho-S473-AKT-specific rabbit anti-human antibody was used (600-401-268, Rockland Immunochemicals, Inc., Gilbertsville, PA, USA). Cells (2.0×104) were treated with or without sorafenib (10 μmol/l), celecoxib (10 μmol/l) or both for 48 h. After washing with PBS, cells were fixed with 4% paraformaldehyde saline. After a further wash with PBS, cells were incubated with Alexa 488-conjugated mouse anti-rabbit IgG antibody (10 μg/ml, Invitrogen, Carlsbad, CA, USA), and incubated for 1 h. Cells were observed under an immunofluoresence microscope (I×81; Olympus).
Intracellular phospho-AKT-specific flow cytometry. Cells were harvested by trypsinization and washed with PBS. For flow cytometric immunostaining, 1×106 cells were fixed on ice in ice-cold 4% paraformaldehyde for 20 min at room temperature, washed once with 1%-BSA-PBS, incubated with Tween (20% in PBS) for 5 min at RT, then washed once again with 1% BSA-PBS. Phospho-AKT expression was determined using flow cytometry (Cytomics FC-500).
Measurement of Prostaglandin-E2 (PGE2) production. HCC cells were treated with 10-40 μmol/l celecoxib, or a combination of 10 μmol/l celecoxib and 10 μmol/l sorafenib, for 48 h. The medium was harvested by centrifugation at 5,000 ×g at 4°C. The amount of PGE2 released into the medium was measured using a specific enzyme immunoassay kit (Cayman Chemical Col, Ann Arbor, MI, USA).
Synergy experiments. Synergy between sorafenib and celecoxib was assessed in HCC cell lines in vitro. Cells were incubated with each agent separately and in combination for 48 h before assessment of cytotoxicity by WST-8 assay as described above. Cells were plated as for WST-8 assays and treated for 48 h with different concentrations of celecoxib and sorafenib, alone and in combination. The half-maximal (50%) inhibitory concentration (IC50) of each drug constituted the median value within the chosen concentration range. Combination index (CI) plots were generated using the CalcuSyn (v2.0) software (Biostat, Cambridge, UK) based on algorithms developed by Chou and Talalay (17) to determine whether the drugs synergize. Based on this approach, CI values <0.9 are considered synergistic, values >1.1 are antagonistic, and values of 0.9-1.1 are approximately additive.
Statistical analysis. All data are expressed as the mean±standard error of the mean. Differences between groups were assessed for statistical significance using the Mann-Whitney test or paired Student's t-test, depending on the distribution of the data. Values of p<0.05 were considered as statistically significant.
Results
Effects of sorafenib-alone and celecoxib-alone on HCC cell growth. To evaluate the effect of sorafenib and celecoxib alone on the growth of the human HCC cell lines, HepG2 and Huh7, analysis of cell growth inhibition was performed by WST-8 assay. The IC50 value, at which growth was 50% of that in the DMSO-treated control, was calculated by non-linear regression analysis using the Excel software. Sorafenib inhibited cell proliferation dose-dependently with an IC50 of 3.4 μM in HepG2 and 4.5 μM in Huh7 cells. Celecoxib also reduced cell viability in a dose-dependent manner with an IC50 of 23.8μM in HepG2 cells and 27.1 μM in Huh7 cells (Figure 1A).
Effects of sorafenib in combination with celecoxib on HCC cell growth. We next examined whether the combination of relatively low concentrations of sorafenib and celecoxib could additively or synergistically inhibit HCC cell growth in vitro. IC10, IC30, and IC60 concentrations of celecoxib were added to HCC cells in the absence or presence of increasing doses of sorafenib (0.01-10 μmol/l) (Figure 1B). Treatment of HepG2 and Huh7 with celecoxib and sorafenib for 48 h resulted in a dose-dependent inhibition of cell viability, as analyzed by WST-8 assay. The time course of cell viability in HepG2 and Huh7 cells after exposure to sorafenib (IC50), celecoxib (IC10), and a combination of these drugs, is shown in Figure 1C.
We further investigated the degree of synergy between these drugs. The addition of celecoxib (at IC30 or IC60) reduced the IC50 for sorafenib to 2.2 μmol/l and 2.1 μmol/l, respectively, in HepG2 cells, and 3.6 μmol/l and 1.7 μmol/l, respectively, in Huh7 cells (Figure 2A). The morphology of HepG2 cells after treatment with sorafenib, celecoxib, and the combination of these molecules is shown in Figure 2B. A CI approach was used to determine whether these molecules caused additive or synergistic inhibition. When celecoxib (IC10, IC30 or IC60) was added to cells incubated with increasing doses of sorafenib, the combinatory effect was evident in both HepG2 and Huh7 cells (Table I). For HepG2 cells, the CI values were between 0.42-0.94. For Huh7 cells, the CI values were between 0.28-0.73, indicating a strong synergism between sorafenib and celecoxib (Table I).
Effects of sorafenib and celecoxib on cell cycle and apoptosis in HCC cells. To investigate whether the sorafenib-mediated reduction of HCC cell viability was caused by suppression of cell division or induction of apoptosis, we analyzed the apoptosis and cell cycle profiles after treatment with sorafenib and/or celecoxib. Treatment with combination of these drugs led to a dramatic increase in apoptotic (sub-G1 phase) cells as shown in Figure 3A. Sorafenib increased the percentage of cells in the sub-G1 phase in a dose-dependent manner (Figure 3B) in both HCC cell lines. At concentrations below its IC50 value, celecoxib did not induce apoptosis of HCC cells (Figure 3B). However, when combined with sorafenib, it significantly elevated apoptosis in both cell lines. CI values for apoptosis induction were between 0.085-0.841 for HepG2 cells, and 0.862-0.988 for Huh7 cells, suggesting synergistic and additive actions, respectively.
The synergy of killing by sorafenib and celecoxib.
Effects of sorafenib and celecoxib on AKT phospholyration in HCC cells. The anti-apoptotic enzyme AKT, has been shown to be an important factor in apoptosis resistance in HCCs (18), and signaling of this enzyme is reduced co-operatively by sorafenib and celecoxib. To ascertain the mechanism of synergy between sorafenib and celecoxib, we examined their effects, alone and in combination, on AKT phosphorylation by flow cytometry. We found that treatment with celecoxib alone and with sorafenib significantly reduced phospho-AKT (Figure 4A). WST-8 assays were performed to further quantify this phospho-AKT (p-AKT) inhibition in HCC cells (Figure 4B). Celecoxib treatment for 24 h reduced the level of phospho-AKT in HCC cells (Figure 4B). In HepG2 cells by the combination of sorafenib and celecoxib, a greater reduction in phospho-AKT was caused than what was observed by either agent alone. A similar trend was observed in Huh7 cells. Finally, we performed immunohistochemical staining of p-AKT in the HepG2 cells as an indication of the activity level of the phosphoinositide 3-kinase (PI3K) signaling pathway, in which AKT is a phosphorylation target. Our data demonstrate a strong expression of phospho-AKT in these cells (Figure 4C).
Effects of celecoxib-alone and in combination with sorafenib on PGE2 production in HCC cells. PGE2 is known to act upon HCCs and its production is up-regulated by COX-2, which is also known to play a role in the induction of AKT phosphorylation. Because celecoxib is a potent COX-2 inhibitor, we next examined the effect of celecoxib and sorafenib on PGE2 production in HepG2 cells. As shown in Figure 5, although high-dose celecoxib inhibits PGE2 production, low doses (IC10-IC30) alone and in combination with sorafenib did not produce such an effect, suggesting that the synergy between celecoxib and sorafenib did not extend to COX-2-dependent PGE2 production in HCC cells. These results suggest that PGE2 production is linked to the inhibition of COX-2, but not to phospho-AKT. Therefore, the synergistic effect of celecoxib and sorafenib in HCC cells appears to be phospho-AKT-dependent but COX-2-independent.

Effect of sorafenib and celecoxib, alone and in combination, on cell viability of human hepatocellular carcinoma (HCC) cells. A: Determination of half-maximal (50%) inhibitory concentration (IC50) of sorafenib-mediated cytotoxicity in HCC cells. Cells were cultured in the presence or absence of sorafenib or celecoxib for 48 h in RPMI-1640 containing 10% FBS. Upper panel: Treatment of HepG2 cells with increasing concentrations of sorafenib (0.001-100 μmol/l; left) and celecoxib (0.001-100 μmol/l; right). Lower panel: Treatment of Huh7 cells with sorafenib (left) and celecoxib (right). Data are expressed as the mean±SEM of five replicates. Data shown are representative of two independent experiments. B: Dose-dependence of the synergistic effect of sorafenib and celecoxib on cell proliferation in HCC cells. HepG2 and Huh7 cells were exposed to different concentrations of sorafenib or/and celecoxib for 48 h. Cell viability was assessed by WST-8 analysis. Data points represent the mean±SEM of three replicate wells within the same experiment. Data sets are: Sorafenib-alone, ●; sorafenib plus an IC10 concentration of celecoxib, ▵; sorafenib plus an IC30 concentration of celecoxib, ⋄; sorafenib plus an IC60 concentration of celecoxib, □. In HepG2 cells, the IC10, IC30 and IC60 values were 10, 15 and 24 μmol/L, respectively. In Huh7 cells, the corresponding values were 11, 18, and 32 μmol/L. C; Time-dependence of treatment effect. Cells were treated with 5 μmol/L of sorafenib alone (●), 10 μmol/l celecoxib alone (○), or a combination of these drugs (▵) for 24, 48, and 72 h, and cell viability was assessed at these time points using the WST-8 assay. Data are expressed as the mean of triplicate measurements. Upper and lower panels show the time dependence of cytotoxicity in HepG2 and Huh7 cells, respectively.
Discussion
In the present study, we have provided evidence that the combination of sorafenib (a RAS/RAF-kinase inhibitor) and celecoxib (a COX-2 inhibitor) leads to potentiated cytotoxicity in the HCC cell lines, HepG2 and Huh7, via increased apoptosis. It appears that in this context, celecoxib is acting on the phosphorylation of AKT downstream of PI3K rather than via COX-2 inhibitory activity. These findings collectively suggest that celecoxib enhances sorafenib-mediated antitumor effects by inhibiting PI3K/AKT signaling in HCC.
The CI values showed that sorafenib and celecoxib administered in combination potentiated cell death signaling in HepG2 and Huh7 HCC cells, particularly at high celecoxib concentrations (IC30, IC60). HepG2 and Huh7 cells differed slightly in their response to celecoxib alone, with HepG2 cells apparently being more susceptible to celecoxib cytotoxicity. However, a higher degree of synergy was obtained when combining a high dose of celecoxib (IC60) with sorafenib in Huh7 cells (CI=0.06) than in HepG2 cells (CI=0.349), although the synergy was appreciable in both cell types. It should be noted that the response to sorafenib in our study differed slightly from the one reported in previous studies on the effect of sorafenib in HepG2 and Huh7 cells (6), which may be accounted for by differences in the sensitivities of the methods used to determine cytotoxicity and apoptosis.
Mechanistically, tumor inhibition by combination therapies may result from an increased capacity of these drugs to induce apoptosis. Sorafenib has been shown to inhibit tumor proliferation and increase apoptosis in HCC (5, 6), and we show here that combining sorafenib and celecoxib potentiates the reduction in cell viability and increase in apoptosis compared to use of either agent alone. Both the RAS/RAF/MEK and the PI3K/AKT signaling pathways promote cell survival, providing a rational for targeting both the RAS/RAF and PI3K pathways (19, 20). Celecoxib, in addition to its more recognized COX-2 inhibitory effects, inhibits PI3K/AKT signaling in cancer (21-23), and we have therefore concluded that the combination of these drugs impinges upon the cellular apoptosis system from two different angles: PI3K-AKT inhibition and RAS/RAF/MEK inhibition (Figure 6).
The role of celecoxib when administered in combination with other drugs in cancer therapy is modulatory rather than therapeutic, and the efficacy of this approach has been reported for various types of cancers (24-26). Celecoxib mediates its antitumor effects by COX-2-dependent and -independent mechanisms (16). Because PGE2 does not seem to be involved in the combination effect of celecoxib with sorafenib in this study, the combination effect of celecoxib appears to be COX-2-independent.
Interestingly, a previous study reported that sorafenib monotherapy stimulated mammalian target of rapamycin (mTOR) and AKT phosphorylation in Huh7 in the presence of EGF (27). In contrast, our study showed that sorafenib inhibits production of phospho-AKT, a difference that may be methodological. Recent evidence hints at cross-talk between the RAS/RAF and PI3K/AKT pathways in a number of cancers (28). Thus, the combination of sorafenib with celecoxib may target both facets of this cross-talk (RAS/RAF and PI3K/AKT) and this dual target approach may underlie the synergistic effects revealed here.
The synergy between sorafenib and celecoxib in HCC cells is clinically significant, because it would be a more cost-effective way of using sorafenib and, perhaps more importantly, because it would lower the potential for adverse events. Sorafenib at clinically effective doses is expensive and causes adverse effects that reduce quality of life, including high rates of diarrhea and HFSR. Although the cost-effectiveness of sorafenib compares favorably with the best supportive care available, it is still unacceptably high when taking into account the cost of treating the adverse events that it causes (29). Reducing the required dose of sorafenib by combining it with celecoxib may diminish both the cost and side-effects associated with its use.

Celecoxib synergism with sorafenib in mediating cytotoxicity in HCC cell lines. A; Celecoxib reduces the IC50 of sorafenib. HepG2 and Huh7 cells were plated for 48 h and treated for a further 48 h with a range of concentrations of sorafenib and celecoxib at 37°C before analyzing cell viability with the WST-8 assay. Data represent the mean±standard deviation of five independently-determined IC50 values. *p<0.05; **p<0.01. B; Morphology of HCC (HepG2) cells after treatment with sorafenib, celecoxib or both.
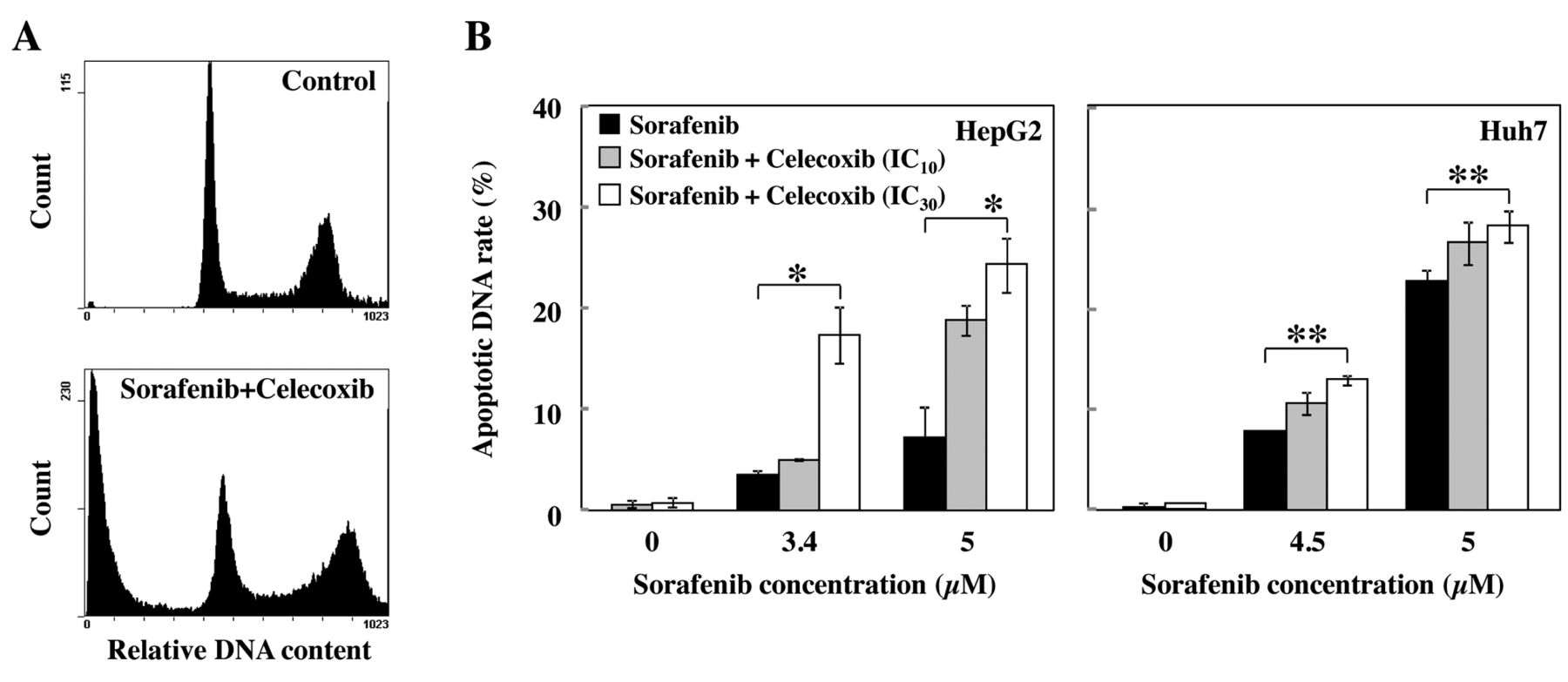
Apoptosis induction by sorafenib and celecoxib, alone and in combination, in cultured HepG2 and Huh7 cells. A; Representative flow cytometric histograms of cells (HepG2) studied with propidium iodide. Cell numbers represent the percentage of cells in the sub-G1 phase of the cell cycle. B; Cells were treated with sorafenib, celecoxib or both for 24 h. Apoptosis was analyzed with flow cytometry. Data show the amount of apoptotic DNA fragmentation under each of these drug treatment conditions (mean±SEM from three replicates). Statistically significant differences from the control (*p<0.05; **p<0.01).
In conclusion, our data show that low-dose sorafenib in combination with celecoxib synergistically inhibits cell viability via actions on the anti-apoptotic AKT and RAS/RAF signaling pathways. Celecoxib potentiates the antiproliferative action of sorafenib, promoting HCC cell apoptosis and allowing for the use of lower doses of sorafenib than those currently used. It may, thus, have potential in treating HCC in patients, a finding that demands further clinical testing.

Flow cytometric analysis of phospho-AKT expression in HCC cells. A; Representative flow cytometric data of phospho-AKT staining in HepG2 (left) and Huh7 (right) cells. B; Semi-quantitation of p-AKT levels in HepG2 and Huh7 cells. Phospho-AKT levels were determined using flow cytometry as described in Materials and Methods. Columns represent the mean±standard deviation of three samples in each group. *Statistically significant difference from the control, sorafenib-alone, or celecoxib-alone (p<0.01). C; Phospho-AKT immunostaining in HCC cells. HepG2 cells were treated with sorafenib, celecoxib, or a combination of these for 48 h, then stained with an antibody against p-AKT (×400).

Effect of sorafenib and celecoxib on prostaglandin-E2 (PGE2) production in HCC cells. HepG2 cells were cultured in a 24-well plate for 24 h in serum-free medium, then treated with sorafenib, celecoxib, or both. Culture medium was then collected, cell debris removed by centrifugation for 20 min at 4°C, and the amount of PGE2 in 100 μl of medium was determined. Columns represent the mean±SD for n=3.

Schematic diagram of the probable mechanism of synergy between celecoxib and sorafenib. RAS/RAF/MAPK pathways and PI3K-AKT pathways control cellular proliferation and apoptosis. The fact that celecoxib and sorafenib act at different points in this signaling network may underpin the synergistic effect seen on the viability of hepatocellular carcinoma cells when exposed to this combination of drugs.
Acknowledgements
The Authors thank the staff of the Cancer Research and Treatment, Graduate School of medical sciences, Kyushu University for provision of cell experimental techniques and for helpful discussions.
Footnotes
-
This article is freely accessible online.
- Received January 16, 2013.
- Revision received February 25, 2013.
- Accepted February 25, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.