


Abstract
Background: The effect of extended exposure of cancer cells to nitric oxide (NO), an endogenous mediator frequently found increased in tumors, is largely unknown. In the present study, the effect of long-term NO exposure on chemotherapeutic resistance was investigated in lung cancer cells. Materials and Methods: The effect of long-term exposure of human lung cancer cells to NO on susceptibility to chemotherapeutic agents-induced apoptosis was analyzed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, co-staining of Hoechst 33342 and propidium iodide (PI), and Annexin V detection. The expression of survival-related proteins was analyzed by western blot. Gene manipulation was used for evaluation of the effect of expression proteins on susceptibility of the cells to chemotherapeutic agent-mediated death. Results: Long-term NO exposure for 7-14 days rendered the lung cancer cells resistant to cisplatin, doxorubicin, and etoposide dose- and time-dependently. The underlying mechanism was found to involve the adaptive responses of the cells, by increasing survival due to increase in the level of caveolin-1 (CAV1) and anti-apoptotic B-cell lymphoma-2 (BCL2), and up-regulation of activated protein kinase B (AKT). The gene manipulation study revealed that the increase of activated AKT and BCL2 was responsible for the resistance to all tested drugs, while the up-regulation of CAV1 only attenuated cell death mediated by doxorubicin and etoposide. Interestingly, NO-mediated drug resistance was found to be reversible when cells were further cultured in the absence of NO for five days. Conclusion: These findings reveal the novel role of NO in the tumor environment, in attenuating chemotherapeutic susceptibility and this could be beneficial in contriving strategies to treat the disease.
Conferred resistance to chemotherapy has become the most interesting issue in the field of cancer research. As found in many types of cancer, an increase of survival pathways of cells facilitates the processes of carcinogenesis, and such pathways remain in malignant cells, potentiating their defenses against anticancer drugs, and increasing their aggressive behaviors (1-4). Strong association was found between augmented survival signals with poor prognosis and resistance to chemotherapy (5-7). In lung cancer, high mortality and low 5-year survival rates were found to be due to two key determinants, drug resistance and metastasis (8, 9). Most combination drugs include those which form DNA adducts, inhibit topoisomerase, and inhibit biosynthesis of macromolecules are frequently prescribed for treatment of lung cancer, however, the efficacy of such therapies was shown to be suppressed by innate (10, 11) or adaptive mechanisms of cancer cells (10, 12). The phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway is a central mediator of cellular survival pathways and has been shown in a number of studies to increase drug resistance and aggressiveness of lung cancer (13-17). Likewise, amplification and overexpression of the B-cell lymphoma-2 (BCL2) proto-oncogene, or other anti-apoptotic members of the BCL2 family of proteins, which occurs in many malignancies, including small cell lung carcinoma, were shown to cause anticancer drug resistance by neutralizing the functions of pro-apoptotic BCL2 family members in apoptotic signaling (18, 19). Recently, evidence has been reported of a significant impact of caveolin-1 (CAV1), a main protein component of caveolae, on various lung cancer cell behaviors, including migration and invasion (20), anoikis resistance (21, 22) and adhesion to endothelial surface (23). Indeed, CAV1 has been shown to be up-regulated in many malignant tumors and linked to poor prognosis (24). Even though the increase of survival signals is accepted as an important mechanism by which cancer cells resist against chemotherapeutic drugs, the molecular insights regarding upstream mediators of such survival augmentations are largely unknown.
Nitric oxide (NO) is widely accepted as one of the most important mediators in cell biology (25-27). NO is synthesized from its substrates, namely L-arginine, NADPH and oxygen, by the catalytic activity of NO synthase enzymes. Studies indicated the presence of inducible NO synthase (iNOS), producing a relatively high level of NO in the micromolar range (28) in many kinds of cancers (29-31). The activity of iNOS in cancer cells was shown to be enhanced in response to pro-inflammatory cytokines (32, 33). Together with evidence indicating that NO is released by immune cells in the tumor area (34), these data support previous findings that this biologically-active molecule plays a significant role in cancer pathology (35-37).
In lung cancer, NOS produces a significant amount of NO in lung neoplasia (38) and elevated NOS activity, as well as increased cancer-related NO production, have been reported in patients with lung cancer (39-41). The effects of NO on lung cancer cells differ, with both anticancer and cancer-promoting effects observed (42, 45). Previously, we found that short-term treatment with NO resulted in the inhibition of BCL2 degradation and thus preserved the cells from Fas ligand (FasL)- (46) and cis-diamminedichloroplatinum (cisplatin)-induced apoptosis (47). On the contrary, NO has been shown to possess pro-apoptotic activity, inducing oxidative stress, caspase activation, and apoptosis (48-52). These controversial findings support the principle that the effect of NO depends on the cell type and cellular redox state, as well as on its concentration and flux (53, 54).
With regard to cancer pathology, cancer cells are surrounded by a microenvironment rich in NO for prolonged periods of time (32, 33, 37). The possible adaptive responses cell to long-term exposure to a low level NO are investigated in the present study. Using pharmacological approaches, we determined the effect of long-term exposure of human lung cancer cells to NO on BCL2 family members and survival proteins, and on susceptibility of cells to cisplatin, doxorubicin, and etoposide-induced apoptosis. Our findings may be important for the explanation of chemotherapy resistance, and might be exploited in cancer chemotherapy.
Materials and Methods
Cells and reagents. NCI-H292 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 100 units/ml penicillin/streptomycin. Cells were trypsinized when cells reached 80-90% confluence. For long-term NO exposure, cells were cultured in medium containing NO donor dipropylenetriamine (DPTA) NONOate (at non-toxic concentrations) for seven and fourteen days. The culture medium was replaced by freshly prepared medium containing DPTA NONOate every two days. The cells were grown in humidified incubators containing an atmosphere of 5% CO2 and 95% air at 37°C. Cisplatin, Hoechst 33342, propidium iodide (PI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO) and annexin V-fluorescein isothiocyanate (FITC) were obtained from Sigma Chemical, Inc. (St. Louis, MO, USA). DPTA NONOate was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Doxorubicin and etoposide were obtained from Calbiochem (San Diego, CA, USA). Antibodies to BCL2, Bcl-2-associated X protein (BAX), Myeloid cell leukemia sequence 1 (MCL1), pAKT, AKT, CAV1, β-actin and peroxidase-labeled secondary antibodies were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). The transfection agent Lipofectamine 2000 was obtained from Invitrogen (Carlsbad, CA, USA).
Plasmid and transfection. CAV1 expression plasmid was obtained from the American Type Culture Collection (ATCC). CAV1, BCL2, and AKT knockdown plasmids (shRNA) were obtained from Santa Cruz Biotechnology. The AKT expression plasmid was obtained from Addgene (Cambridge, MA, USA). The BCL2 plasmid was generously provided by Dr. C. Stehlik (West Virginia University Cancer Center, Morgantown, WV, USA). Stable transfections of overexpression, control, or knockdown plasmids of CAV1, AKT, and BCL2 were generated by culturing H292 cells in a six-well plate until they reached 60% confluence. Fifteen microliters of Lipofectamine 2000 reagent and 2 μg of overexpression, control, or shRNA plasmids were used to transfect the cells in the absence of serum. After 12 h, the medium was replaced with culture medium containing 5% FBS. Approximately 36 h after the beginning of transfection, the cells were digested with 0.03% trypsin, plated onto 75-cm2 culture flasks, and cultured for 24 to 28 days with antibiotic selection. The stable transfectants were pooled, and the expression of CAV1, AKT, and BCL2 proteins in the transfectants were confirmed by western blotting. The cells were cultured in antibiotic-free RPMI-1640 medium for at least two passages before use in each experiment.
Cell viability assay. Cell viability was determined by the MTT assay. Cells were seeded in a 96-well plate and allowed to attach for 12 h. Cells were treated with different concentrations of DPTA NONOate (0-200 μM) for 24, 48 and 72 h, and cells were incubated with 500 μg/ml of MTT at 37°C for 4 h. Then, the MTT solution was removed and 100 μl of DMSO was added to dissolve the formazan crystals. The intensity reading of MTT product was measured at 570 nm using a microplate reader, and the percentage of viable cells was calculated relative to that of control cells.
Nuclear staining assay. Apoptotic and necrotic cell death were determined by Hoechst 33342 and PI co-staining. After specific treatments, cells were incubated with 10 μM of Hoechst 33342 and 5 μg/ml of PI for 30 min at 37°C. Nuclei condensation and DNA fragmentation of apoptotic cells and PI-positive necrotic cells were visualized and scored under a fluorescence microscope (Olympus IX51 with DP70; Olympus, Center Valley, PA, USA).
Annexin V detection by flow cytometry. Apoptosis was determined by annexin V-FITC staining assay. Treated and untreated cells were collected, re-suspended, and incubated with annexin V-FITC for 30 min at room temperature, and cells were scored as apoptosis by flow cytometry using a 488-nm excitation beam and a 538-nm band-pass filter (FACSort; Becton Dickinson, Rutherford, NJ, USA). The mean fluorescence intensity was quantified by CellQuest software (Becton Dickinson).
Western blot analysis. After specific treatments, cells were incubated in lysis buffer containing 20 mM Tris-HCl (pH 7.5), 1% Triton X-100, 150 mM sodium chloride, 10% glycerol, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 100 mM phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN, USA) for 40 minutes on ice. The cell lysates were collected, and the protein content was determined using the Bradford method (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of proteins from each sample (60 μg) were denatured by heating at 95°C for 5 min with Laemmli loading buffer and subsequently loaded onto 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel for electrophoresis. After separation, proteins were transferred onto 0.45 μM nitrocellulose membranes (Bio-Rad, CA, USA). The transferred membranes were blocked for 1 h in 5% nonfat dry milk in TBST (25 mM Tris-HCl pH 7.5, 125 mM NaCl, and 0.05% Tween 20) and incubated with the appropriate primary antibodies at 4°C overnight. The membranes were then washed twice with TBST for 10 min and incubated with horseradish peroxidase-labeled isotype-specific secondary antibodies for 2 h at room temperature. The immune complexes were detected by enhancement with chemiluminescence substrate (Supersignal West Pico; Pierce, Rockfore, IL, USA) and quantified using analyst/PC densitometry software (Bio-Rad).
Statistical analysis. Mean data from at least three independent experiments were normalized to the results for the control cells. Statistical differences between means were determined using an analysis of variance (ANOVA) and post hoc test at a significance level of p<0.05, and data are presented as the mean±SD.
Results
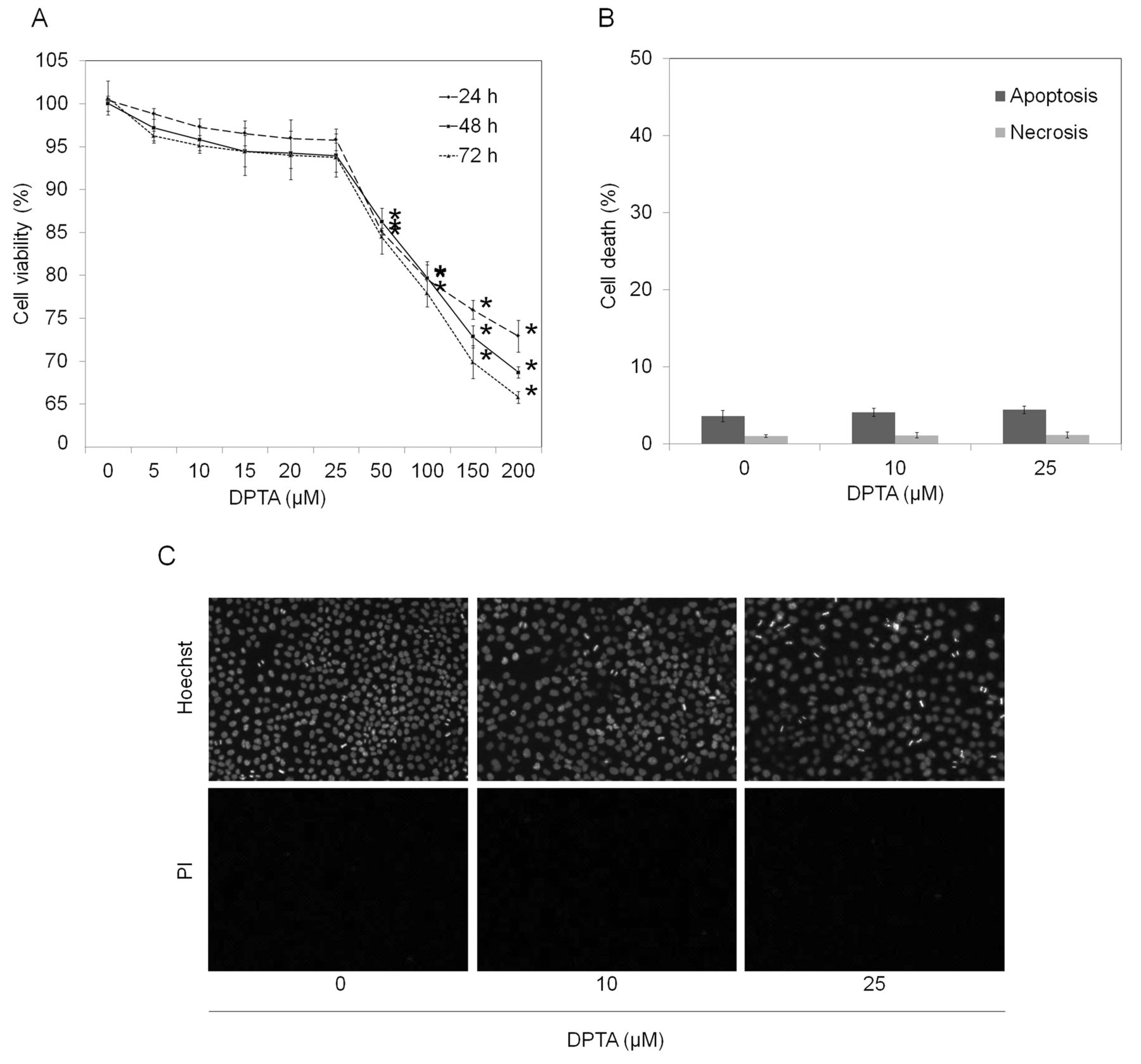
Effect of NO donor, DPTA NONOate, on cell viability of human lung cancer cells. We first characterized the effects of NO donor on cell viability and selected concentrations that caused no significant effects on the viability of the cells for further experiments. Cells were cultured in the presence or absence of DPTA NONOate (5-200 μM), a slow-releasing NO donor, and cell viability was determined after 24-72 h. Results showed that treatment of the cells with DPTA NONOate at 5-25 μM caused neither cytotoxicity nor proliferative effects on the cells (Figure 1A). A significant decrease of cell viability was first detected in cells treated with 50 μM for 24 h, when approximately 85% of the cells remained alive. The concentrations of NO donor at 10 and 25 μM were selected and their non-toxic effect was confirmed by apoptosis and necrosis detection. Figure 1B and C show that treatment of the cells with 10 and 25 μM caused no significant alteration in terms of apoptosis and necrosis in comparison to those of the non-treated control.
Long-term NO exposure potentiates cell resistance to chemotherapeutic agents. Chemotherapy resistance has been long recognized as an importance obstacle to successful cancer therapy (7). The widely prescribed drugs for lung cancer treatment, cisplatin, doxorubicin and etoposide, were used in the present study. Lung cancer H292 cells were cultured in the presence or absence of NO donors for seven and fourteen days, as described in Materials and Methods. They were then treated with different concentrations of the drugs for 24 h and viability, apoptosis, and necrosis were determined. Figure 2A indicates that cisplatin at 25-100 μM significantly reduced cell viability of H292 cells in a dose-dependent manner, with an approximately 40% reduction in cell viability observed in the 100-μM cisplatin-treated group. Interestingly, the cells exposed to DPTA NONOate at 10 and 25 μM exhibited significant resistance to such cisplatin-mediated death (Figure 2A). It is worth noting that the NO-treated cells in both experiments exhibited similar drug-resistant profiles. Treatment of the cells with cisplatin also caused a dose-dependent increase in cell apoptosis over that of the controls, as indicated by increased nuclear fluorescence and chromatin condensation detected by Hoechst 33342 and PI co-staining. At 50 μM cisplatin, approximately 40% of H292 cells exhibited apoptotic morphology, whereas the PI-positive necrosis cells were undetected. Interestingly, NO treatment significantly reduced the number of apoptotic cells induced by cisplatin treatments (Figure 2B and C). To confirm the apoptosis-inducing effect of cisplatin, cells were similarly treated with cisplatin, and apoptotic cells were determined by annexin V-based assay and flow cytometry. Consistently, our results showed that cisplatin-induced apoptosis in NO-treated H292 cells was significantly reduced in comparison to that of the control H292 cells (Figure 2D). Similar treatments, as well as cell viability, apoptosis, and necrosis evaluations were applied for investigating the effects of long-term NO treatment on sensitivity to doxorubicin and etoposide. For doxorubicin, treatment of the H292 cells with 1-5 μM of the drug resulted in a significant decrease of cell viability (Figure 3A). As expected, exposure to NO donors for seven and fourteen days at 10 and 25 μM significantly reduced the response of the cells to doxorubicin. The dose-dependent effect of NO treatment on doxorubicin could only be detected in the cells that were treated with NO for seven days, whereas at 14 days, similar resistance was observed at both concentrations of NO. Likewise, apoptosis assays (Hoechst 33342 and annexin-V) confirmed that the NO-treated cells exhibited a significant reduction of apoptosis in response to doxorubicin treatment compared that of the control cells (Figure 3B and D). In accordance with the above findings, treatment with NO significantly reduced susceptibility of cells to etoposide-mediated death. Figure 4 indicates that cells treated with NO for seven and fourteen days exhibited a significant decrease in cellular response to etoposide-mediated death in comparison to their parental H292 cells. Taken together, our observations have shown the negative regulatory effect of extended NO treatment on cell susceptibility towards these chemotherapeutic agents.

Effect of dipropylenetriamine (DPTA) NONOate on cell viability of human lung cancer H292 cells. Cells were treated with different concentrations of DPTA NONOate (0-200 μM) for 24, 48 and 72 h. A: Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. B: Apoptosis and necrosis cell death were determined by Hoechst 33342 and propidium iodide (PI) co-staining assay. C: Apoptotic and necrotic cells were visualized under a fluorescence microscope. Values are means±SD, (n=3); *p<0.05 versus non-treated control cells.
NO mediates chemotherapy resistance in lung cancer cells by increasing cellular survival. Having shown that NO exposure induces cell resistance to cisplatin, doxorubicin, and etoposide, the present study further identifies the possible underlying mechanisms. Focusing on the fact that patients with lung cancer suffer from low responses after receiving combination regimen for treatment, the activation and up-regulation of survival pathways were proposed as an important cause. While activated AKT has garnered the most attention and was observed to be up-regulated in a number of resistant cancer cells (13), increasing evidence has indicated the prominent role of anti-apoptotic proteins namely, BCL2 and MCL1, on chemoresistance (55, 56).
We, thus, characterized the survival-related protein profiles of 7- and 14-day NO-treated H292 cells and compared them against those of H292 cells without NO. Figure 5 shows that the total AKT level in NO-treated and control H292 cells were comparable and not affected by either dose or length of time of treatment. The significant increase of phosphorylated AKT at Ser473 (activated form) was observed in the NO-treated cells in comparison to that of the control cells. This NO-mediated AKT activation was found to be in a concentration-dependent manner, and further increase was observed when the duration of NO exposure was prolonged. Likewise, the expression of anti-apoptotic BCL2 protein was found to exist strongly up-regulated in NO-treated cells. NO exposure at 10 and 25 μM induced BCL2 up-regulation in dose- and time-dependent manners, with an approximately 3-fold induction observed when the cells were treated with 25 μM of NO donor for 14 days. The expression levels of CAV1 were shown to be increased in response to NO exposure, whereas NO treatment caused no significant effect on the levels of anti-apoptotic MCL1 and pro-apoptotic BAX.
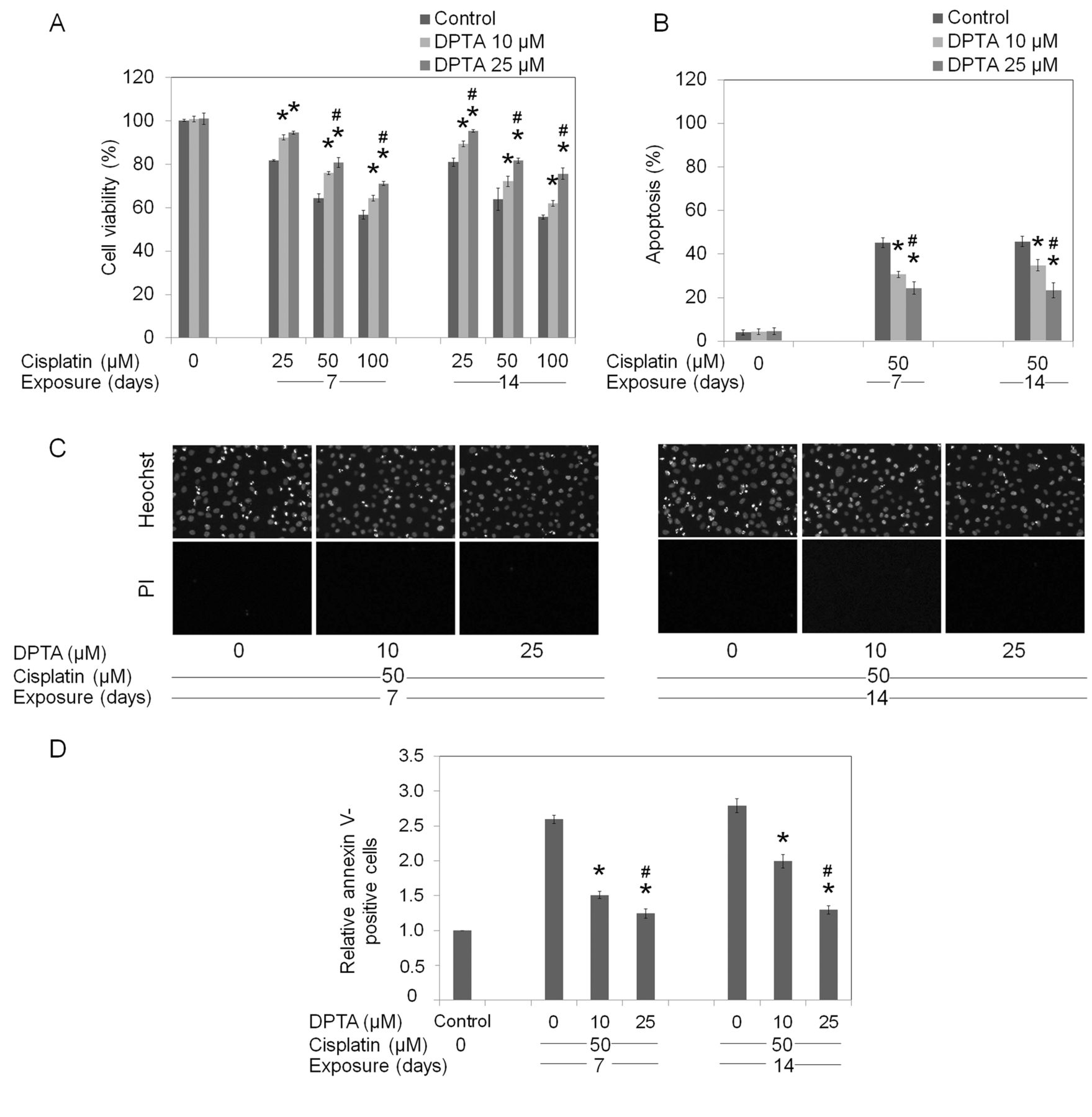
Long-term dipropylenetriamine (DPTA) NONOate exposure mediates cell resistance to cisplatin. H292 cells were incubated with DPTA NONOate (0-25 μM) for either 7 or 14 days, prior to treatment with cisplatin (0-100 μM) for 24 h. A: Cell survival was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. B: Apoptotic cell death was determined by Hoechst 33342 and propidium iodide (PI) co-staining assay. C: Morphology of cell nuclei was visualized under a fluorescence microscope after Hoechst 33342/PI co-staining. D: Apoptosis was evaluated by flow cytometry using annexin V-fluorescein isothiocyanate (FITC). Values are means±SD, (n=3); *p<0.05 versus non-treated cells, #p<0.05 versus 10 μM DPTA-treated cells.
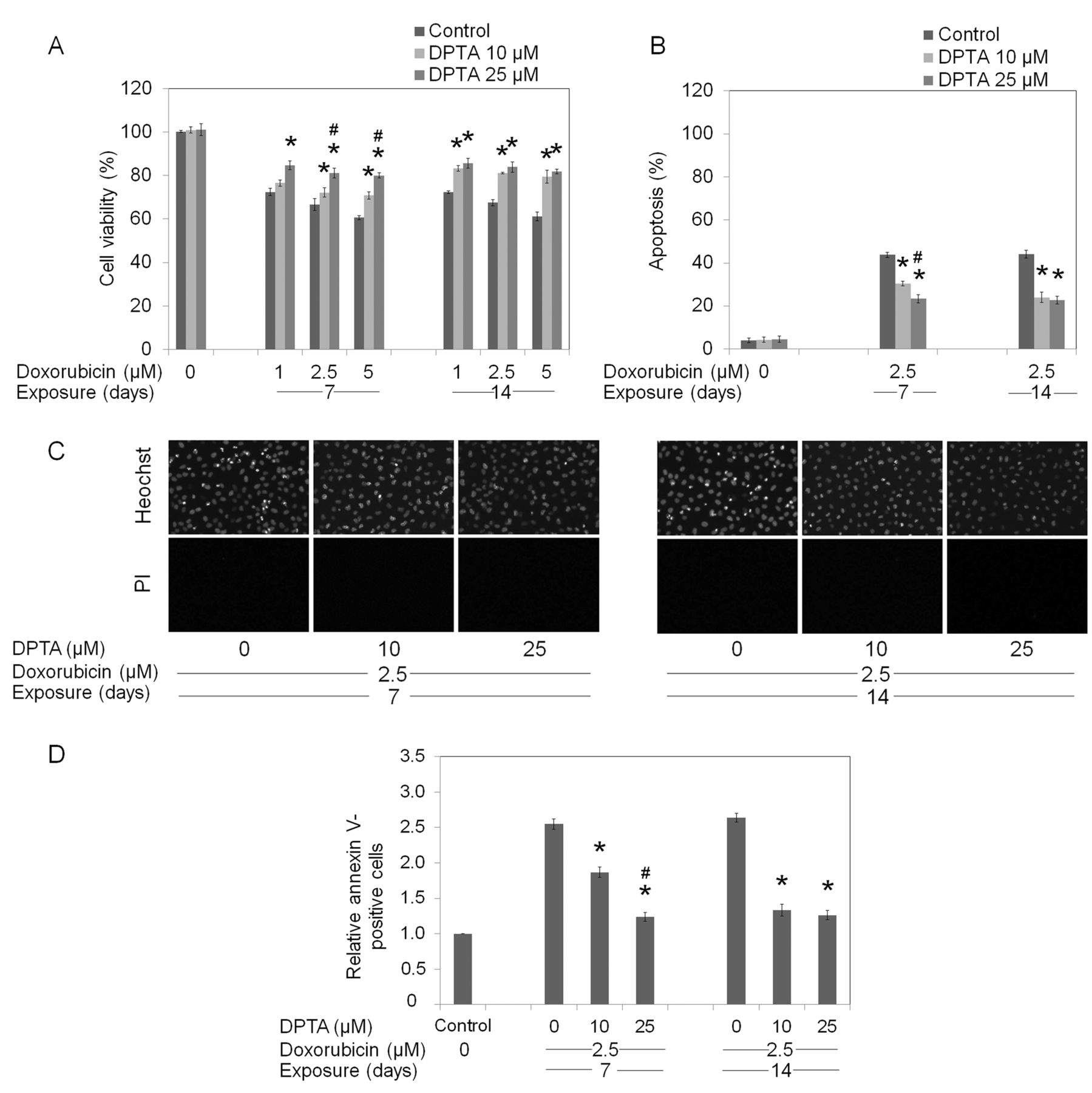
Long-term dipropylenetriamine (DPTA) NONOate exposure mediates cell resistance to doxorubicin. H292 cells were incubated with DPTA NONOate (0-25 μM) for either 7 or 14 days, prior to treatment with doxorubicin (0-5 μM) for 24 h. A: Cell survival was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. B: Apoptotic cell death was determined by Hoechst 33342 and propidium iodide (PI) co-staining assay. C: Morphology of cell nuclei was visualized under a fluorescence microscope after Hoechst 33342/PI co-staining. D: Apoptosis was evaluated by flow cytometry using annexin V-fluorescein isothiocyanate (FITC). Values are means±SD, (n=3); *p<0.05 versus non-treated cells, #p<0.05 versus 10 μM DPTA-treated cells.
To test whether the increases in survival-related proteins are involved in resistance to apoptosis induced by cisplatin, doxorubicin, and etoposide, we stably transfected the cells with overexpressing or shRNA plasmids of CAV1, AKT, and BCL2 and we determined the cell viability after chemotherapeutic drug treatments. Figure 6A shows that cells transfected with CAV1-, AKT-, and BCL2-overexpressing plasmids exhibited up-regulation of their corresponding proteins, while the ShRNA-transfected counterparts showed significantly decreased levels. To determine the effect of each protein on the susceptibility of the cells to drug-mediated death, stably-transfected cells were left untreated or treated with 50 μM cisplatin, 2.5 μM doxorubicin, or 25 μM etoposide, and viability was measured at 24 h. MTT-based cell viability assay indicated that CAV1-overexpressing cells exhibited significant resistant to doxorubicin and etoposide but with no effect on cisplatin. In addition, shCAV1 only significantly sensitized cells to etoposide-mediated death. For AKT-transfected cells, the increase of activated AKT significantly attenuated the death response of the cells to all three drugs. The shAKT-transfected cells exhibited increased susceptibility to doxorubicin- and etoposide-mediated toxicity. Accordingly, cells overexpressing BCL2 exhibited resistance to all three drugs and the shRNA knock-down of the protein significantly increased the response of the cells to cisplatin, doxorubicin, and etoposide. Taken together, these results suggest that the increase of CAV1 levels in response to NO rendered cells resistant to doxorubicin and etoposide but not cisplatin. However, the increase of activated AKT and BCL2 in response to long-term NO could render cells resistant to all three drugs.

Long-term dipropylenetriamine (DPTA) NONOate exposure mediates cell resistance to etoposide. H292 cells were incubated with DPTA NONOate (0-25 μM) for either 7 or 14 days, prior to treatment with etoposide (0-50 μM) for 24 h. A: Cell survival was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. B: Apoptotic cell death was determined by Hoechst 33342 and propidium iodide (PI) co-staining assay. C: Morphology of cell nuclei was visualized under a fluorescence microscope after Hoechst 33342/PI co-staining. D: Apoptosis was evaluated by flow cytometry using annexin V-fluorescein isothiocyanate (FITC). Values are means±SD. (n=3); *p<0.05 versus control cells, #p<0.05 versus 10 μM DPTA-treated cells.
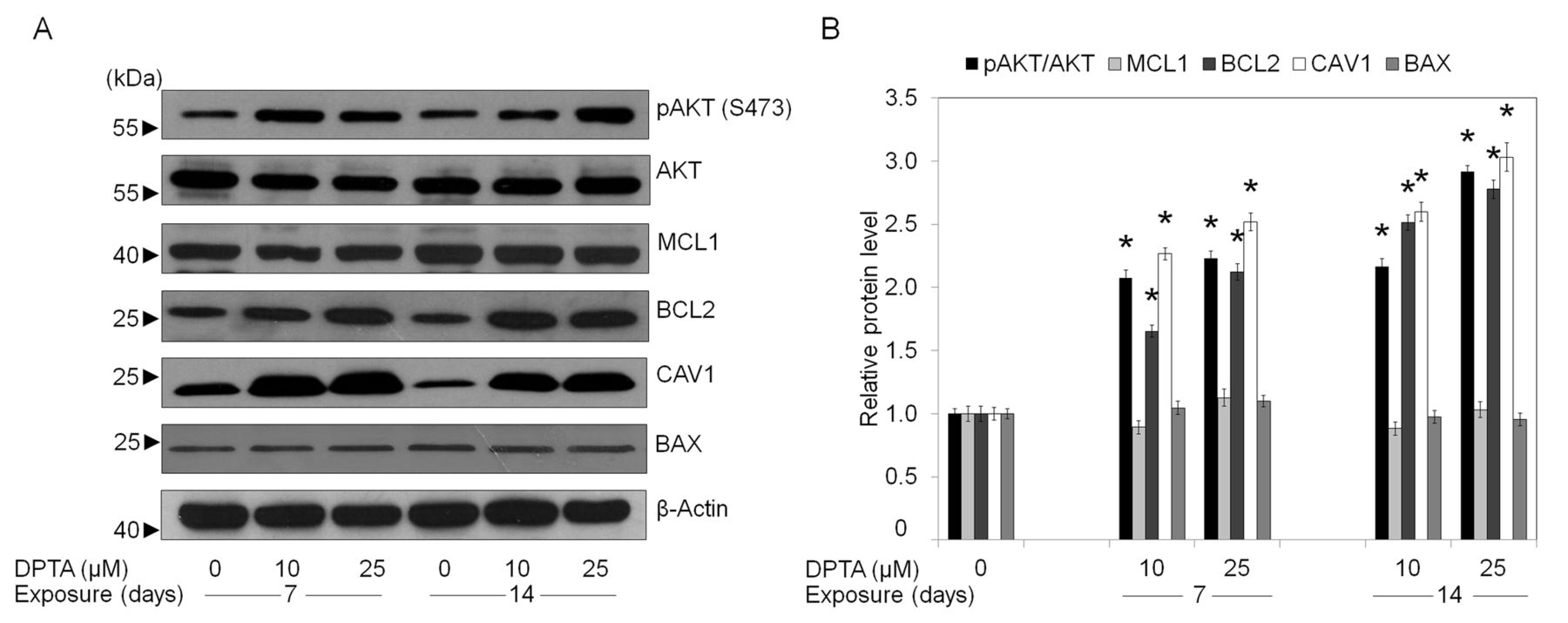
Nitric oxide influences cellular survival proteins. A: After H292 cells were incubated with dipropylenetriamine (DPTA) NONOate (0-25 μM) for 7-14 days, cells were collected and analyzed for the expression of phosphorylated protein kinase B (AKT) (Ser 473), total AKT, B-cell lymphoma-2 (BCL2), Myeloid cell leukemia sequence-1 (MCL1), Bcl-2-associated X protein (BAX) and caveolin-1 (CAV1) proteins by western blotting. Blots were reprobed with a β-actin antibody to confirm equal loading of samples. B: Immunoblot signals were quantified by densitometry and mean data from independent experiments were normalized by blot of the controls. Values are means±SD, (n=3); *p<0.05 versus non-treated control cells.

Effect of caveolin-1 (CAV1), protein kinase B (AKT), and B-cell lymphoma-2 (BCL2) proteins on susceptibility of H292 cells to cisplatin-, doxorubicin-, and etoposide-mediated death. A: H292 cells were stably transfected with overexpressing, control, or shRNA plasmids of CAV1, AKT, or BCL2 and the levels of proteins were analyzed by western blot. B: Stably-transfected cells were treated with 50 μM cisplatin, 2.5 μM doxorubicin, or 25 μM etoposide for 24 h and cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Values are means of samples±SD, (n=3); *p<0.05 versus H292 control cells.
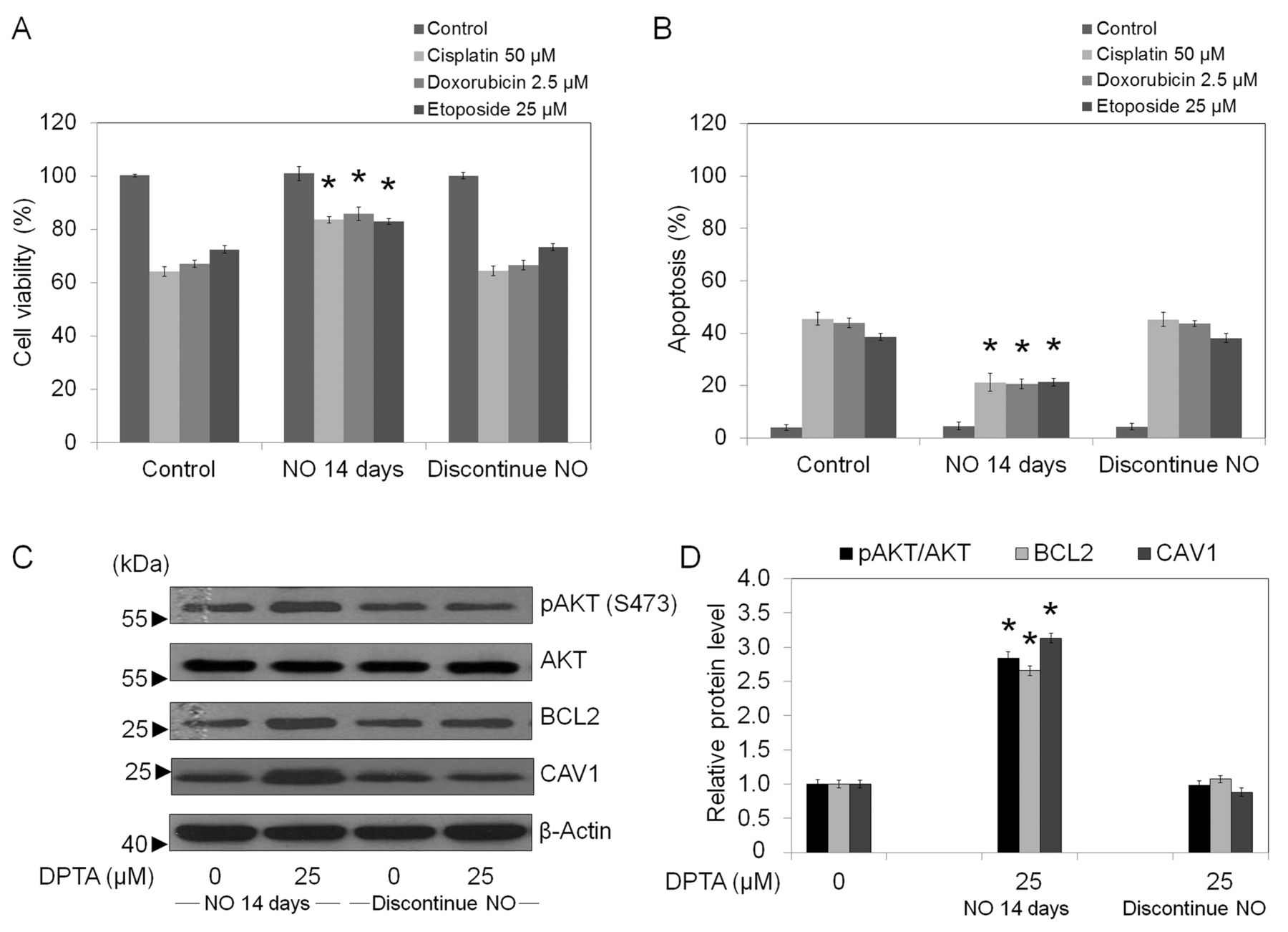
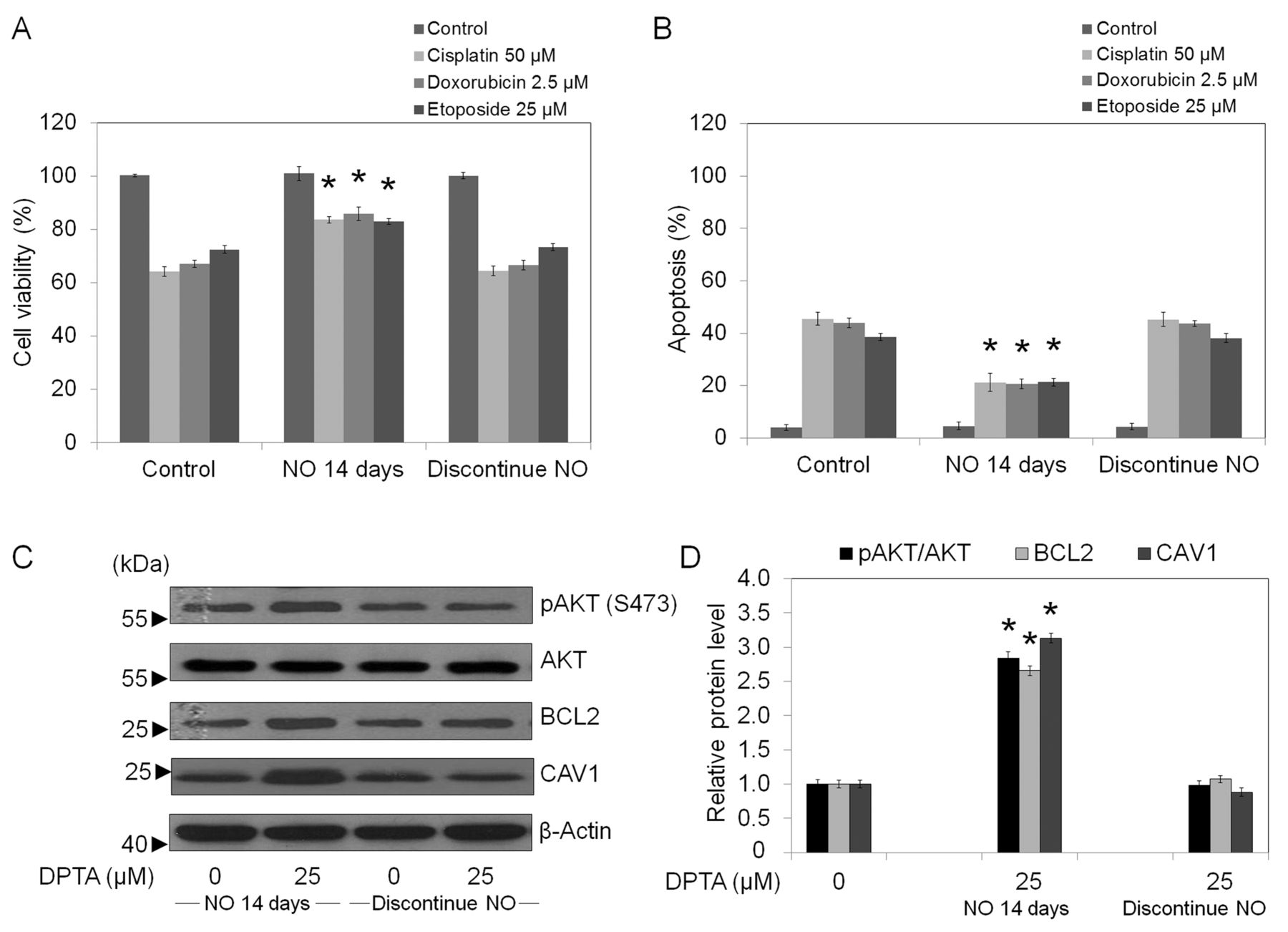
Effect of NO discontinuation on chemotherapeutic resistance. The NO-treated H292 cells (14 days) were cultured in the absence of NO donor for five days. The cells were then treated with cisplatin (50 μM), doxorubicin (2.5 μM), or etoposide (25 μM) for 24 h. A: Cell survival was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Data represent means±SD. (n=3); *p<0.05 versus drug-treated control cells. B: Apoptotic cells were evaluated by Hoechst 33342 staining. Data represent means±SD. (n=3); *p<0.05 versus drug-treated control cells. C: Cells were collected and analyzed for CAV1, AKT, pAKT, and BCL2 expressions by western blotting. Blots were re-probed with a β-actin antibody to confirm equal loading of samples. D: Immunoblot signals were quantified by densitometry, and mean data from independent experiments were normalized to the results for the controls. Data represent mean±SD. (n=3); *p<0.05 versus H292 control cells.
The effect of NO-mediated chemoresistance is reversible. Having shown that up-regulation of survival-related proteins was responsible for chemotherapy resistance of NO-treated cells, we next examined whether these effects could be reversed in the absence of NO donors. The cells after 14 days of NO exposure were cultured in the absence of NO for 5 days. The cells were then tested for their chemotherapy susceptibility and the expression levels of CAV1, AKT, pAKT, and BCL2 were determined using western blotting. Figure 7A and B show that cell viability reverted to the base line level when the cells were grown under NO-free conditions. Notably, such a reversal was observed as early as three days after NO discontinuation (data not shown) and the reversal was steady, reaching the level of the control cells at five days. In addition, the expressions of CAV1, pAKT, and BCL2 were found to be comparable to those of the control cells (Figure 7C and D).
Discussion
Mechanisms involved in cancer-related drug resistance have been proposed and demonstrated through several studies. Among them, the attenuation of apoptosis has garnered the most recognition (57). Several pathways are suggested to be involved in such chemoresistance, including an up-regulation of anti-apoptotic members of the BCL2 family (55), an increase of antioxidants and related detoxifying mechanisms (58), and the enhancement of survival signals of the cells (13). Since such resistance occurred even during the use of combination drugs acting on different cellular targets, survival augmentation seems to be a possible mechanism for cancer cells resistance to various death stimuli.
Accumulated clinical observations have supported the above concepts by pointing out a significant increase of cell survival in a variety of human cancer types (4, 5, 13, 14) and such mechanisms are linked to chemotherapy resistance (5, 13). An increase of survival signals in cancer cells was shown to be mediated through AKT activation, playing the role of a core regulator. While increased activity of AKT caused by gene mutations was found in many types of cancer, including breast and lung cancers (59), the mechanism of up-regulation of AKT activity by acquired means is largely unknown.
We and others hypothesized that endogenous substances found surrounding the tumor including reactive oxygen species (ROS), NO, and pro-inflammatory cytokines, can force cells to change their behaviors (60) or phenotypes (61). Our observations in the present study indicated that NO-mediated drug resistance can be reversed when the cells are cultured under NO-free conditions, with cell sensitivity being restored to baseline levels at five days of NO discontinuation (Figure 7). These results suggested that the cellular responses involving pAKT, BCL2, and CAV1 up-regulation are due to the adaptive machinery of the cells. Results from this study also support the concept that NO present in the cancer microenvironment plays a possible role in regulation of chemotherapy resistance.
In terms of DNA-interacting agents, such as cisplatin and doxorubicin, cell-cycle arrest at the G2/M phase was shown to be a critical event necessary for apoptosis induction (62). Studies indicated that the induction of the AKT pathway inhibits apoptosis by reducing the levels of p21 and p27, inhibitors of cyclin-dependent kinases, and prevents the down-regulation of cyclins A, B, and E (63). In addition, an attenuated apoptosis of cancer cells by an increase in the cellular level of anti-apoptotic proteins has been extensively reported (64). Along with the evidence already mentioned, the present study shows that NO triggered the resistance to chemotherapeutic drugs by an increase in AKT function. We also found that NO exposure significantly increases the level of anti-apoptotic BCL2 protein, while having only minimal effect on MCL1 protein (Figure 5). Previously, we found that short-term NO exposure (12 h) rendered H460 human lung cancer cells resistant to cisplatin-mediated death (47). In that study, NO S-nitrosylated BCL2 and prevented the protein from proteasomal degradation. Even though it is possible that such S-nitrosylation may have up-regulated BCL2 protein in the present study, S-nitrosylated proteins in fact have a short half-life (65).
With gene manipulation, we generated stable transfectants of H292 cells exhibiting different levels of CAV1, BCL2, and AKT protein expressions, in order to verify the functions of each protein in drug resistance. The results indicated that BCL2, as well as pAKT, up-regulation in the H292 cells conferred cisplatin, doxorubicin, and etoposide resistance. However, the up-regulation of CAV1 in these cells inhibited only doxorubicin- and etoposide-mediated death. This finding is in accordance with the previous study indicating that CAV1 has less effect on cisplatin-mediated death and in certain studies CAV1 was found to sensitize cells to cisplatin-induced apoptosis (66).
In summary, the present study has provided evidence indicating that NO present in the tumor microenvironment has a significant impact on cell susceptibility to chemotherapeutic agents by increasing the expression of survival regulatory proteins. The findings of the present work could be important for the development of novel cancer therapies and encourages further studies in related areas.
Acknowledgements
The Authors would like to thank the 90th Anniversary of Chulalongkorn University, Ratchadaphiseksompot Endowment Fund, Chulalongkorn University (RES560530132-HR), and Mr. Krich Rajprasit and Mr. Leonard Fordham, proofreaders.
- Received September 30, 2013.
- Revision received November 3, 2013.
- Accepted November 5, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}