


Abstract
Background: Shortened, constitutively active androgen receptor (AR) isoforms have been characterized and linked to tumor progression and chemoresistance in prostate cancer (PCa). We examined the regulation of shortened AR isoforms by a newly-identified AR regulatory signaling pathway involving heat-shock protein HSPB1 and microRNA miR-1. Materials and Methods: HSPB1 and miR-1 were modulated by overexpression and knock-down approaches utilizing the model PCa system, 22Rv1. Subsequently, AR isoform expression levels were quantified by western blot analysis. Results: HSPB1 was identified as an inducer and miR-1 as an inhibitor of AR variants, with no detectable discrimination between long and short AR isoform regulation. Conclusion: In 22Rv1 cells, all AR isoforms were co-regulated by the cytoprotective factor HSPB1 and the tumor suppressor miR-1. Notably, our data provide evidence that HSPB1 inhibition is able to target expression of long as well as of short AR isoforms.
The androgen receptor (AR) is a member of the nuclear receptor family and is mainly activated by testosterone and dihydrotestosterone. Beyond control of prostate development and physiology, the AR is an essential modulator of prostate cancer (PCa) genesis and progression. Concerning this central role in tumor biology, AR functionality is currently targeted by antihormonal PCa therapy inhibiting androgen synthesis, as well as AR activity. Previous studies discovered shortened AR isoforms in human PCa cell lines and tissues, which were suspected of causing resistance to anticancer therapy (1-5). Mutations, alternative splicing and proteolytic cleavage have been proposed to generate different AR isoforms (3, 6, 7). However, most of these isoforms share the common structural features of variable, truncated COOH-terminal regions and hence lack the ligand-binding domain of the receptor. Therefore, shortened AR proteins have been proposed to contribute to constitutive, ligand-independent transcriptional activity, and, subsequently, to enhanced cellular growth and tumor progression (3, 4, 8). A new generation of drugs to treat PCa are small molecules specifically targeting co-factors of AR functionality, e.g. clusterin (9), heat-shock protein 90α and 90β (HSP90AA1 and HSP90AB1) (10, 11), 52 kDa FK506-binding protein (FKBP4) (12), and finally the small heat-shock protein HSPB1 (heat-shock protein-27) (13). Recently, we found AR expression be controlled by a regulatory cascade of the cytoprotective factor HSPB1 and the tumor-suppressor microRNA-1 (miR-1) (14, 15). To assess effects of the putative signaling axis HSPB1–miR-1–AR on shortened AR isoforms, we analyzed the well-established PCa cell line 22Rv1, which was derived from a xenograft model of PCa that relapsed during antihormonal therapy (4, 16). 22Rv1 cells express a long variant of AR protein (long AR; 120 kDa) genetically determined by an in-frame duplication of exon 3 (4), and, more importantly, at least two constitutively active AR isoforms (short AR; ~80 kDa) caused by COOH-terminally deleted ligand-binding domains (17). In this study, we investigated the role of the newly-identified AR modulators HSPB1 and miR-1 on the expression of shortened AR isoforms in a model PCa system.
Materials and Methods
Cell culture. Human PCa cell lines LNCaP, 22Rv1 and PC-3 were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were propagated in RPMI 1640 medium with 10% fetal bovine serum and 1% penicillin/streptomycin (PAN Biotech, Aidenbach, Germany) at 5% CO2 atmosphere and 37°C. One day before dihydrotestosterone (DHT; Sigma-Aldrich, Deisenhofen, Germany) treatment, cells were plated into 6-well cell culture plates (150,000 cells/well). Incubation experiments were carried out in RPMI 1640 media without phenol red with 1% penicillin/streptomycin and fetal bovine serum replaced by 5% Panexin NTA (PAN Biotech) and 10 nM DHT. Control cells were treated with ethanol as vehicle.
Transfection experiments. One day before transfection, cells were plated into 6-well cell culture plates (150,000 cells/well). For modulation of HSPB1 and miR-1, cells were transfected using Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) with a total amount of 5 μg plasmid DNA or using siLentFect (BioRad, München, Germany) with 40 nM Anti-hsa-miR-1 miScript miRNA Inhibitor (Qiagen, Hilden, Germany), respectively. Empty vectors pcDNA3.1 (Invitrogen) and pSuperior (OligoEngine, Seattle, WA, USA) served as control.
Cloning of pAR. The pAR plasmid encoding for full-length AR cDNA sequence was generated by amplifying the coding sequence of the plasmid pSG5-AR (kindly provided by M. Cronauer, Ulm, Germany) utilizing specific oligonucleotides AR-1 FOR (5’-AAAAAGGATCCATGGAAGTGCAGTTAGGGC-3’) and AR-2763 REV (5’-TTTTTCTCGAGTCACTGGGTGTGGAAATAG-3’). The PCR product was ligated into the BamHI/XhoI restricted multiple cloning site of the expression vector pcDNA3.1 (Invitrogen). Plasmid clones were verified by sequencing.
Cloning of pshHSPB1. The DNA plasmid pshHSPB1 encoding for an HSPB1-specific siRNA cDNA sequence was constructed by using the pSuperior vector system from OligoEngine. Two oligonucleotides shHSPB1-534 FOR (5’-GATCCCCGATCACCATCCCAGTCACCTTCAAGAGAGGTGACTGGGATGGTGATCTTTTTA-3’) and shHSPB1-534 REV (5’-AGCTTAAAAAGATCACCATCCCAGTCACCTCTCTTGAAGGTGACTGGGATGGTGATCGGG-3‘) were hybridized by a temperature gradient from 95°C to 4°C for 40 min. Due to the asymmetrical design of both complementary oligonucleotides, the hybridization products formed defined single-stranded 5’ overhangs which were ligated into the BglII/XhoI digested vector pSuperior. Plasmid clones were verified by sequencing.
Western blotting analysis. Cells were harvested in lysis buffer (50 mM Tris (pH 7.5), 150 mM NaCl, 10 mM K2HPO4, 5 mM EDTA, 10% glycerol, 1% Triton X-100, 0.05% sodium dodecysulfate, 1 mM Na3VO4, 20 mM NaF, 0.1 mM phenylmethylsulfonyl fluoride, 20 mM 2-phosphoglycerate) and a complete protease inhibitor cocktail from Roche Applied Science (Mannheim, Germany). Protein concentrations were determined using the Bradford Reagent (BioRad) and equal amounts of total protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After transfer to a nitrocellulose membrane (GE Healthcare Europe, Freiburg, Germany), western blots were blocked (Rotiblock; Carl Roth, Karlsruhe, Germany) and incubated with primary antibodies directed against HSPB1, AR, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cell Signaling Technology, Danvers, MA, USA) overnight. Subsequently, membranes were incubated with a peroxidase-coupled secondary antibody (Cell Signaling Technology) for 1 h and proteins were visualized by utilizing SuperSignal West Dura Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA) in a ChemiDoc XRS+ System (BioRad). Quantification of protein signals was performed by Image Lab 3.0 software (BioRad).
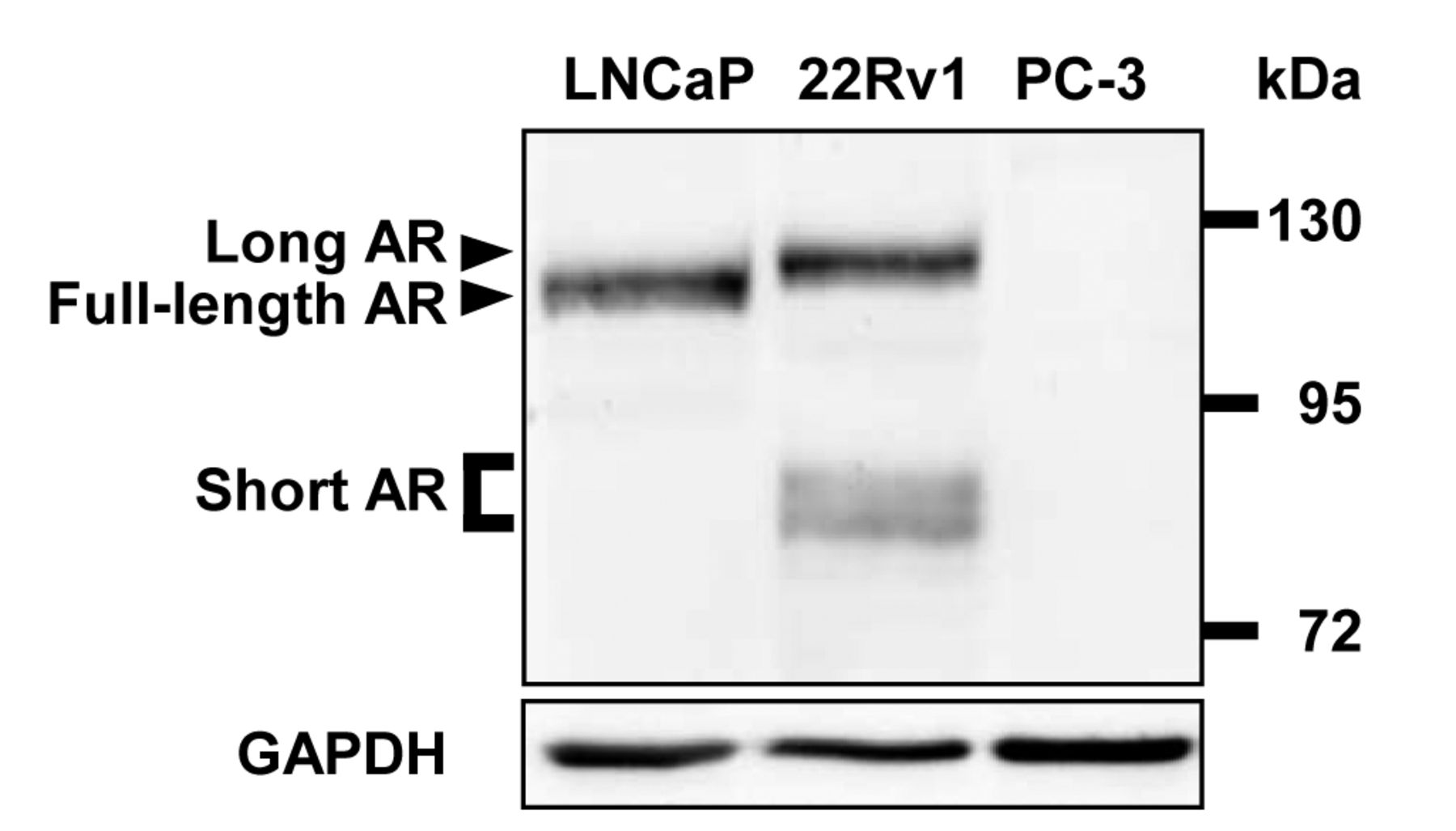
Androgen receptor (AR) isoforms in prostate cancer cell lines LNCaP, 22Rv1, and PC-3. Equal amounts of total cell lysates were analyzed by western blotting utilizing an AR-specific antibody directed against the NH2-terminus of AR protein. Detection of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as loading control.
MicroRNA quantification by quantitative reverse transcription polymerase chain reaction (RT-PCR). MicroRNA was prepared applying the mirPremier microRNA isolation Kit (Sigma-Aldrich). Subsequently, reverse transcription was performed with 2 μg of total microRNA and the miScript II RT Kit (Qiagen) followed by real-time PCR quantification using miScript SYBR Green PCR Kit (Qiagen), Hs_miR-1_1 miScript Primer Assay (Qiagen) and the CFX96 Real-Time PCR Detection System (BioRad). Hs_RNU6-2_1 miScript Primer Assay (Qiagen) served as reference.
Statistical analysis. All experiments were repeated at least four times. Statistical analysis was performed using the unpaired Student's t-test. Values of p≤0.05 were considered significant.
Results
Expression of shortened AR isoforms is stimulated in the presence of DHT and by AR autoinduction. To verify our experimental cell model system we initially examined the AR expression status in the PCa cell lines LNCaP, 22Rv1, and PC-3 (Figure 1). As expected, LNCaP cells expressed full-length AR (115 kDa), whereas western blot analysis of 22Rv1 cells revealed protein signals of long AR (120 kDa) and at least two shortened isoforms (~80 kDa), hereafter called short AR. PC-3 cells were negative for AR protein (18). DHT stimulation of 22Rv1 cells led to weak but significant induction of long AR (1.3-fold; p=0.010) and short AR (1.2-fold; p=0.028) compared to vehicle-treated control cells (Figure 2A and B). Subsequently, autoinductive properties of AR were assessed utilizing the expression vector pAR. Overexpression of full-length AR protein increased the expression rate of long AR (3.7-fold; p=0.002), as well the one as of short AR (2.2-fold; p=0.046; Figure 2C and D), and confirmed the functionality of the established model cell system.

Autoinduction of long and short androgen receptor (AR) isoforms by dihydrotestosterone (DHT) and overexpression of wild-type AR. A: 22Rv1 cells were treated with 10 nM DHT for 48 h. Levels of long and short AR were determined by western blotting using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as loading controls. B: Densitometric quantification of long and short AR expression normalized to vehicle-treated controls. C: 22Rv1 cells were transfected with wild-type AR-encoding plasmide pAR. After 48 h, levels of long and short AR were determined by western blotting using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as loading control. D: Densitometric quantification of expression of long and short AR normalized to expression of cells transfected with control vector pcDNA3.1.
The cytoprotective heat-shock protein HSPB1 is an inducer of long AR and short AR expression. Recently, we found that full-length AR expression is regulated by HSPB1 (14). Therefore, western blot analyses of 22Rv1 cells overexpressing HSPB1 were conducted and indicated expression of long AR and short AR to be significantly higher than in controls (long AR 1.5-fold; p=0.025; short AR 1.4-fold; p=0.049; Figure 3A and B). We studied whether knock-down of HSPB1 caused inhibitory effects on AR isoforms. For this, pshHSPB1 plasmid expressing an HSPB1-specific small-interfering RNA was generated as described in the materials and methods section and applied in transfection experiments. Confirming the overexpression approaches mentioned above, suppression of HSPB1 in 22Rv1 cells caused a decrease of levels of AR isoforms levels (long AR: 0.5-fold; p=0.046; short AR: 0.6-fold; p=0.011; Figure 3C and D).
Expression of long and short AR is blocked in the presence of tumor-suppressive miR-1. The small regulatory microRNA miR-1 is a newly-identified negative regulator of AR expression (15). These data prompted us to study whether miR-1 contributes to modulation of shortened AR expression. Similar to HSPB1 studies mentioned above, we modulated miR-1 expression by a DNA plasmid encoding for miR-1 mimicking RNA (pmiR-1) (15) and by a commercially available miR-1-specific microRNA inhibitor. Prior to AR analysis, efficacy of miR-1 overexpression and knock-down was confirmed by miR-1-specific quantitative RT-PCR (data not shown). As shown in Figure 4, elevated levels of miR-1 were accompanied by reduced concentrations of AR isoforms (long AR: 0.5-fold, p=0.002; short AR: 0.4-fold, p=0.013; Figure 4A and B). On the other hand, experimental abrogation of miR-1 effects by inhibition resulted in up-regulation of long and short AR (long AR: 1.2-fold, p=0.009; short AR: 1.6-fold, p=0.011; Figure 4C and D). Again, these findings attested to co-regulation of long and short AR by the putative signaling axis HSPB1–miR-1 in 22Rv1 cells.
Discussion
In this study, we demonstrated that the COOH-terminally shortened variants of AR in 22Rv1 cells, short AR, are induced by the cytoprotective factor HSPB1, that short AR expression is attenuated by the HSPB1-regulated tumor-suppressor miR-1, and most notably, that long and short AR isoforms are not differentially regulated by the signaling axis HSPB1–miR-1.
A set of factors contributing to AR expression levels in PCa cells has been tested. After transfection of full-length AR, the induction of long AR was found to be weakly but significantly higher than the induction of short AR (Figure 2D). This effect was most likely caused by an overlay of protein signals of transfected full-length AR and endogenously induced long AR due to very similar molecular weights of both proteins (full-length AR: 115 kDa; long AR: 120 kDa). Furthermore, incubations with DHT (Figure 2B) again demonstrated equally-induced expression of long and short AR proteins by autoinduction. More interestingly, there was no statistically significant evidence for differential regulation of AR isoforms, neither by HSPB1 nor by miR-1. As expected, long and short AR were comparably induced by HSPB1 and suppressed by miR-1, indicating co-regulation of these AR isoforms by the recently identified signaling axis HSPB1–miR-1. Targeting HSPB1 by the newly developed nucleic acid-based drug OGX-427 offers new and potent therapy options for advanced PCa (13, 19), however, there is little known regarding the potential effects of HSPB1 inhibition on shortened AR isoforms. Besides suppression of general cytoprotective effects (20), inhibition of HSPB1 reduces AR levels (14) and subsequently eliminates proliferative and thereby pro-oncogenic signals in PCa progression. Importantly, our data provide evidence that HSPB1 inhibition is able to target long as well as short AR isoforms. This is even more relevant since COOH-terminal shortened AR isoforms are ligand-independent and constitutively active, and therefore most likely resistant to antagonistic drugs and inhibitors of androgen biosynthesis (21). Consequently, HSPB1 inhibition therapy is potentially beneficial even to patients with advanced PCa caused, at least partially, by expression of shortened AR variants, and therefore supports clinical treatment with HSPB1-targeting anticancer therapeutics.
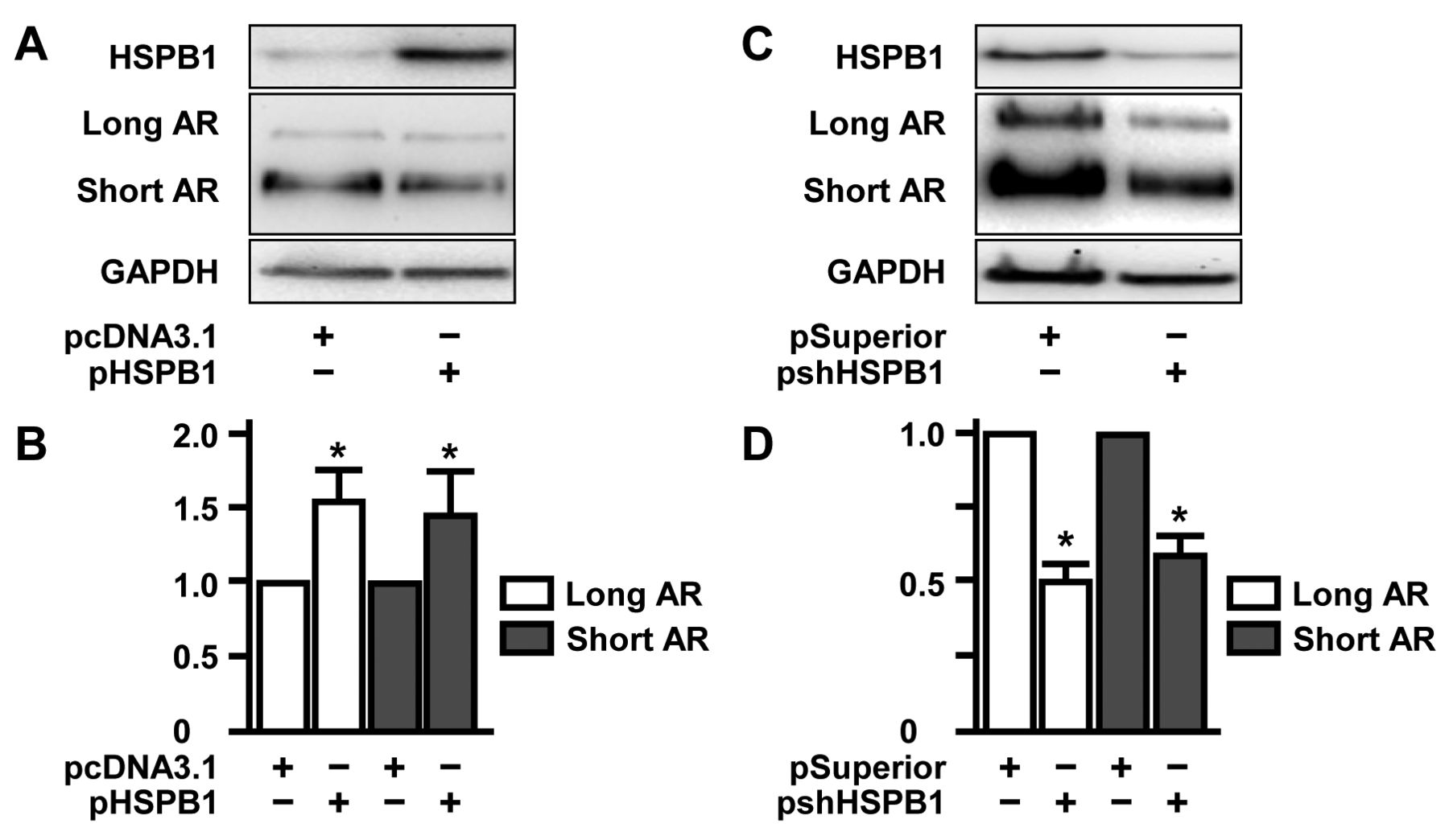
Induction of long and short androgen receptor (AR) isoforms by the cytoprotective factor heat-shock protein HSPB1. A: 22Rv1 cells were transfected with the HSPB1-encoding plasmid pHSPB1. After 48 h, expression levels of HSPB1 as well as long and short AR were determined by western blotting using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as loading control. B: Densitometric quantification of expression of long and short AR normalized to that of cells transfected with control vector pcDNA3.1. C: 22Rv1 cells were transfected with the plasmid pshHSPB1 encoding for an HSPB1-specific siRNA cDNA sequence. After 48 h, expression levels of HSPB1 as well as long and short AR were determined by western blotting using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a loading control. D: Densitometric quantification of expression of long and short AR normalized to that of cells transfected with control vector pSuperior.
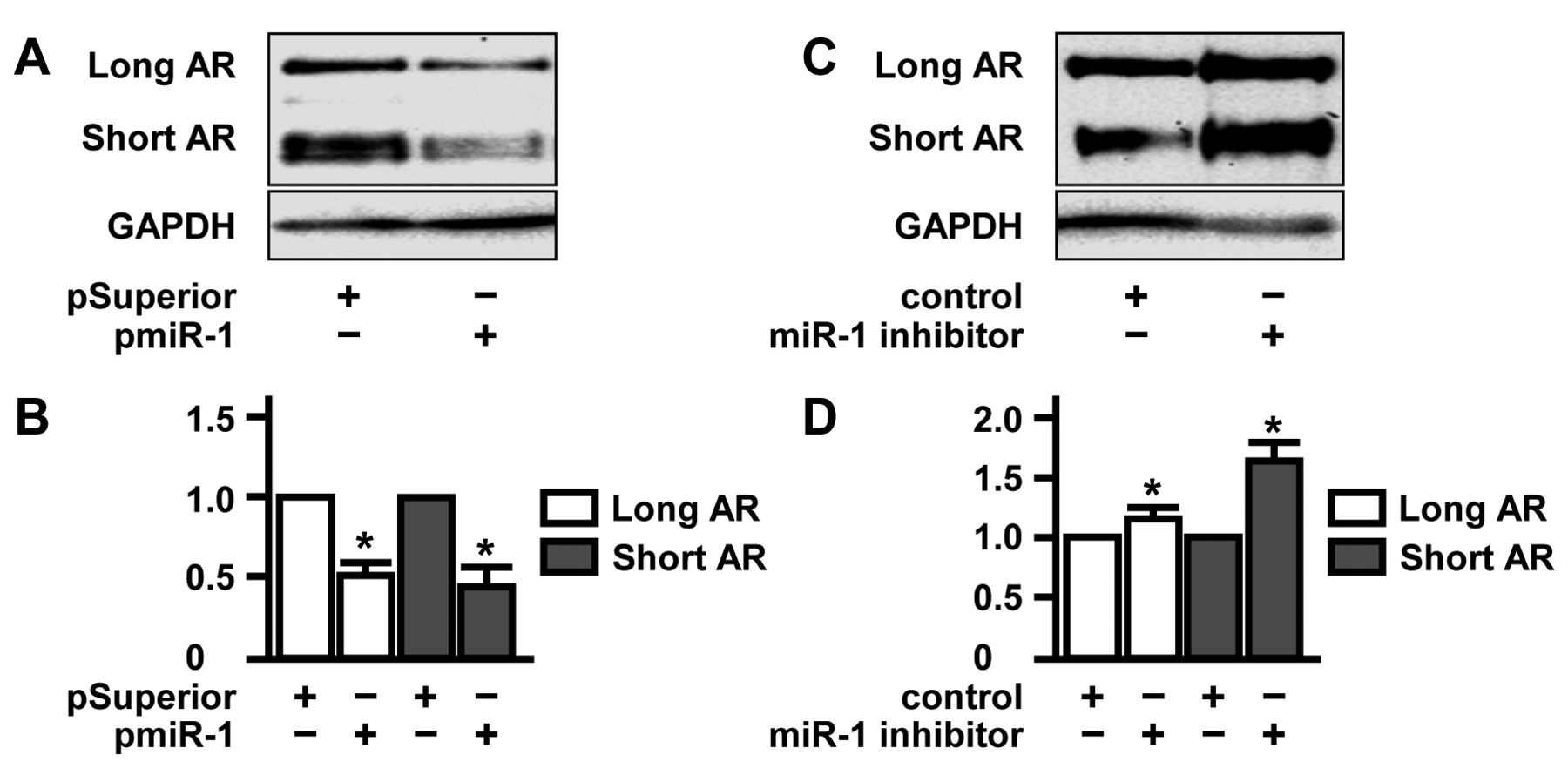
Suppression of long and short androgen receptor (AR) isoforms by the tumor-suppressor microRNA miR-1. A: 22Rv1 cells were transfected with the plasmid pmiR-1 encoding for an miR-1-mimicking cDNA sequence. After 48 h, expression of long and short AR was determined by western blotting using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a loading control. B: Densitometric quantification of expression of long and short AR normalized to that of cells transfected with control vector pSuperior. C: 22Rv1 cells were transfected with 40 nM Anti-hsa-miR-1 miSript miRNA Inhibitor (Qiagen, Hilden, Germany). After 48 h, expression of long and short AR levels was determined by western blotting using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as loading control. D: Densitometric quantification of expression of long and short AR normalized to that of control transfected cells.
Acknowledgements
The Authors thank Anne Brandenburg and Katja Wittig for her excellent technical assistance. Parts of this work were supported by a grant of the Doktor Robert Pfleger-Foundation, Bamberg, Germany.
- Received September 23, 2013.
- Revision received October 17, 2013.
- Accepted October 18, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Reinforcement of the Tumor Suppressing Properties of microRNA-1 by Substitution at the C2' Position of Varying Ribose Residues in Chemically Synthesized microRNA-1 Molecules
- Physiological and Genetically Engineered Expression Modulation Methods Do Not Affect Cellular Levels of the Heat Shock Protein HSP60 in Prostate Cancer Cells
- Gelsolin Governs the Neuroendocrine Transdifferentiation of Prostate Cancer Cells and Suppresses the Apoptotic Machinery
- Overexpression of MicroRNA-1 in Prostate Cancer Cells Modulates the Blood Vessel System of an In Vivo Hen's Egg Test-Chorioallantoic Membrane Model
- New Treatment Options for Osteosarcoma - Inactivation of Osteosarcoma Cells by Cold Atmospheric Plasma
- Heat-shock Protein HSPB1 Attenuates MicroRNA miR-1 Expression Thereby Restoring Oncogenic Pathways in Prostate Cancer Cells