


Abstract
Aim: Colorectal cancer (CRC) is one of the most common malignant diseases. The aim of our study was to describe the expression status of 12 selected candidate genes, by comparing paired samples of healthy colon mucosa and tumour tissues and to correlate obtained data with clinical and pathological features, with the goal of revealing associations for individual gene expressions and tumour behaviour. Materials and Methods: Samples from 53 patients with CRC were analyzed. Patients were divided into two groups based on the presence or absence of distant metastases at the time of primary tumour surgery. Expression levels were assessed by quantitative real-time polymerase chain reaction. Results: We found changes in the expression of 10 out of 12 analyzed genes. Four genes were significantly up-regulated in tumour tissues: leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5; p<0.001), collagen triple helix repeat containing 1 (CTHRC1; p<0.001), visinin-like 1 (VSNL1; p<0.001) and versican (VCAN; p=0.001). Six genes were down-regulated: destrin (DSTN; p=0.004), mesoderm induction early response 1, family member 3 (MIER3; p<0.001), acyl-CoA synthetase long-chain family member 5 (ACSL5; p=0.002), mitogen-activated protein kinase 1/ERK (MAPK1; p<0.001), claudin 23 (CLDN23; p<0.001) and solute carrier family 26 (sulfate transporter), member 2 (SLC26A2; p<0.001). We recorded longer overall survival (OS) in the group of patients with higher expression of VSNL1 (p=0.032). Patients with more pronounced down-regulation of CLDN23 had shorter OS (p=0.045). In the group of patients without distant metastases, longer OS and disease-free interval (DFI) were found for patients with higher SLC26A2 expression in tumour tissues (p=0.036 and p=0.011, respectively). In the same group, lower expression of VSNL1 in healthy tissue corresponded to a longer DFI (p=0.020), smaller decrease of SLC26A2 and ACSL5 meant longer DFI (p=0.041 and p=0.040, respectively), as did greater increase of LGR5 expression (p=0.026). Conclusion: We identified differences in the expression of 10 genes in colorectal cancer tissue compared to healthy colon mucosa, and found prognostic significance for these changes which could be used for the development of a disease risk scoring system.
The incidence of colorectal cancer (CRC) is increasing and globally, this malignant disease has the third highest incidence (after breast and lung cancer) and the fourth highest mortality rate (after lung, liver and stomach cancer) (1). In the first stages of its development, CRC can be treated by surgical intervention. In later stages, it has a high capacity to form secondary tumours, mainly in the liver and lungs (2). Therefore it is neccessary to combine surgery and chemotherapy to achieve higher efficacy of treatment.
The selection for optimal treatment is also complicated by the high heterogeneity of CRC, which can be divided into three main subtypes: type with chromosomal instability, type with microsatellite instability and type with CpG island methylator phenotype (3). Individual subtypes differ in disease prognosis and prediction, and their identification is often crucial for the effective eradication of residual disease by oncological treatment.
Currently, there are several chemotherapeutic regimens and the possibility of biological treatment also exists. The most commonly used chemotherapeutics are 5-fluorouracil, oxaliplatin and irinotecan, usually in various combinations (4). Biological treatment is targeted against the epidermal growth factor receptor (EGFR) or vascular endothelial growth factor receptor (VEGFR). Two categories of agents acting against EGFR exist: small tyrosine kinase inhibitors (gefitinib, erlotinib) and monoclonal antibodies (panitumumab and cetuximab) (5). The most complicated step in the treatment is the selection for the appropriate agent for each patient individually.
Summary of genes selected for the study.
At present, only a few predictive and prognostic markers are used in CRC therapy and several others are in the phase of experimental validation. Two of these predictive markers are related to the use of biological treatment, the expression status of the EGFR and the mutational status of the EGFR proximal effector, the small G-protein Kirsten rat sarcoma viral oncogene homolog (KRAS) (6, 7). Only patients who have tumours positive for EGFR expression and have a wild-type allele for the KRAS gene can be treated by anti-EGFR treatment, but there is still a substantial proportion of patients who meet the elegibility criteria but lack treatment benefit (8). Other markers are being studied, namely the mutational status of other RAS family genes, v-raf murine sarcoma viral oncogene homolog B (BRAF) and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) or the expression of phosphatase and tensin homolog (PTEN) protein (negative regulator of the PI3K pathway) (9, 10). Markers predicting the efficacy of classical chemotherapy are also studied, for example the excision repair cross-complementing rodent repair deficiency, complementation group 1 (ERCC1) polymorphisms in the oxaliplatin treatment (11) or DNA topoisomerase 1 (TOP1) expression level in irinotecan treatment (12). Markers with prognostic function are of particular interest because on their potential to inform about disease agressiveness and for their possible contribution to follow-up optimization.
Despite the number of studied genes, there is still a need to identify for novel markers whose mutation or expression status would provide additional information that would be useful for more precise patient selection, with the final goal of individualized medicine. Therefore, here, we have selected potentially interesting candidate genes according to their known or possible function in diverse aspects of tumour progression, i.e. angiogenesis, metabolism or cell adhesion, and tried to relate their altered expression with clinical behaviour of primary CRC.
Materials and Methods
Selection of studied genes. The selection of studied genes was based on a search through the dataset from the high-throughput studies focused on colorectal cancer (13, 14). Our aim was to find genes whose expression change was already identified in the large-scale analysis, but was never confirmed by different approaches. Our search finally narrowed down 12 candidate genes whose names and identificators are summarised in Table I.
Two of the genes - leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5) and mitogen-activated protein kinase 1/ERK (MAPK1) - were selected as control genes with known and verified expression change in CRC. Selection of reference genes glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and polymerase (RNA) II (DNA directed) polypeptide A (POLR2A) was based on the data from our collaborators (15) and our own laboratory practice.
Primer design. Quantitative real-time polymerase chain reaction (PCR) primers for the selected genes were designed using Primer-3 software (16) with focus on having similar annealing temperatures and lengths of the PCR product. Primer sequences are listed in Table II. Primers were synthetised by Sigma Aldrich Company (St. Louis, MO USA) using their custom oligo synthesis service.
Collection of tissue samples. Samples for the analysis were collected during surgery for the colorectal tumour. Within 20 min after the removal of the tumour tissue from the patient, small samples of tumour and healthy mucosa (anatomically the most distant tissue which was macroscopically healthy, usually in the range of 15-20 cm from the tumour location) were collected. Samples were immediatelly frozen in cryotubes (Thermo Fisher Scientific, Waltham, MA, USA) and stored at −80°C.
List of primer sequences, annealing temperatures and amplicon lengths. tm, Melting temperature; a.l., amplicon length.
Isolation of RNA and quality validation. RNA was isolated from frozen samples using the standard protocol for the RT Trisol Reagent (Molecular Research Center, Cincinnati, OH, USA). The tissue was removed from the cryotube and placed in liquid nitrogen in a mortar. The tissue was pulverized and transferred into an Eppendorf tube with 1ml of chilled Trisol RT and total RNA was isolated according to the manufacturer's protocol. Total isolated RNA was dissolved in nuclease-free water (Ambion, Carslbad, CA, USA). RNA concentration was assessed by absorbance measurement using the Infinite M200 (Tecan, Männedorf, Switzerland) in the NanoQuant setting. Only samples with a 230 nm/260 nm ratio >1.7 and samples with proper bands without degradation on agarose gel electrophoresis were used for further analysis. Selected samples were additionally analysed by measurement of RNA integrity number (RIN) using a 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA).
Reverse transcription (DNase, PCR control). cDNA was synthetised using 500 ng of total RNA in 20 μl reaction by RevertAid First Strand cDNA Synthesis Kit (Fermentas, Waltham, MA, USA) according to the manufacturer's protocol. Before reverse transcription, isolated RNA was treated for 5 min at room temperature with DNaseI (Top-Bio, Vestec, Czech Republic) to remove potential traces of genomic DNA. For priming of reverse trancription, we used a combination of oligo(dT)18 and random hexamer primers each at 2.5 μM final concentration. The quality of cDNA and possible contamination by genomic DNA was assessed by control PCR reaction (GAPDH amplification, 40 cycles) and agarose gel electrophoresis.
Quantitative real-time PCR. For the quantitative PCR we used Power SYBR Green PCR master mix (Life Technologies, Carslbad, CA, USA). cDNA was diluted to final concentration of 0.5 ng/μl and 4 μl of cDNA were used in each reaction. Optimal cycling parameters and annealing temperatures were assessed by the measurement of sensitivity, specificity and efficiency of individual quantitative PCR reactions. After this initial analysis, genes were divided into two groups based on their ideal annealing temperature (58°C for CLDN23, SLC26A2, VSNL1, CAPN10, VCAN and MAPK1; 60°C for LGR5, DSTN, MIER3, ACSL5, CTHRC1 and SAMD3). The instrument used for the analysis was 7500 Fast (Life Technologies). Cycling parameters were: initial hold at 50°C for 20 s and initial denaturation at 95°C for 10 min followed by 42 cycles consisting of denaturation at 95°C for 15 s and annealing and polymeration at 58°C (60°C) for one minute. Results were analysed by the 7500 instrument software and basic statistical analysis was carried out using the REST2009 software (Qiagen, Hildesheim, Germany).
Collection of clinical information. Clinical data were retrieved from the patient's records. We focused on the pathological examination of the samples (pTNM classification, grading and histological type of tumour) and on the patient's data (gender, age, date of diagnosis and surgical intervention and date of the last follow-up examination, recurrence, death). All data were anonymised.
Statistical analysis. Since the normality of the measured expression levels is not certain, nor can it be reliably tested given the limited sample size, non-parametric statistical methods, which do not require any assumption regarding the distribution shape, were preferred during the analysis.
First the differences in the candidate genes expressions between tumorous and healthy tissue were tested for significance using the Wilcoxon signed-rank test.
Then a series of two-sample survival analyses was performed in order to investigate possible relations between the expression data (healthy tissue expression level, tumorous tissue expression level and tumorous/healthy tissue expression ratio for each candidate gene) and patient survival (calculated from the day of surgery). The two patient groups to be compared were formed independently for each variable based on its median value. In cases where patients were later removed from the analysis due to missing data or when only a subset of patients was analyzed, the median value used to divide patients into two groups was kept at its original value. The relation between the expression variables and overall survival (OS) time was analyzed for all the patients and also in the subsets of adjuvantly and palliatively treated patients separately. In the group of patients treated adjuvantly, the correspondence between the expression data and the disease-free interval (DFI) after surgery was also investigated. The date of disease recurrence was determined as the average of the date of the last negative and the first positive examination if the interval between the examinations was 180 days or less. In cases of a longer examination interval, the recurrence date was set 90 days before the first positive examination based on previous methodology (17). During each analysis, Kaplan-Meier curves were plotted for the two patient groups based upon the variable of interest and the significance of the survival (or DFI) difference was tested by Gehan-Wilcoxon, Cox-Mantel and “log-rank” test.
Clinical and pathological description of the analysed group.
To examine the possible relation between tumour localisation and candidate gene expression, the patients were first divided into two groups according to tumour localization (first group with the CRC of the colon, second group with CRC of the rectum and rectosigmoid). The candidate gene expression levels (in both tumour and healthy tissue) and expression ratios were then tested for significant differences between the groups using Mann-Whitney U-test.
Correlations of expression variables were investigated using Spearman's correlation coefficient with appropriate significance test.
All the analyses were performed in STATISTICA (StatSoft, Tulsa, OK, USA).
Results
In the present study, we analysed the expression of 12 genes in a sample set of 53 patients. Patients were divided into two groups, palliative and adjuvant (25 and 28 patients, respectivelly), depending on the presence or absence of distant macrometastases at the time of surgery. Clinical and pathological description of both groups is summarised in Table III. Both groups differed clinically in overall survival, with patients of the adjuvant group having longer survival than those in the palliative group.
Different RNA expression between tumour tissue and healthy mucosa. In the set of 12 selected genes, 10 exhibited a difference in expression level between tumour and normal tissue in the group of all patients. Four (LGR5, CTHRC1, VSNL1 and VCAN) were up-regulated in tumour, six (DSTN, MIER3, ACSL5, MAPK1, CLDN23 and SLC26A2) were down-regulated (Figure 1). Expression differences in both subgroups were similar to those obtained for the whole set of patients, except for DSTN, the expression of which was not different in the palliative group, but was down-regulated in the adjuvant group.
Correlation between candidate gene expressions and clinical data. Dependence of clinical characteristics on relative expression levels of candidate genes was statistically analyzed by the tests described above. We did not find any significant result at a p-value of 0.05 for correlation with age, individual parameters of TNM classification and tumour grading.

Expression of candidate genes in tumour tissue relative to healthy tissue. Genes in dark grey were up-regulated in tumour tissue, genes in light grey did not change in expression and genes in white were down-regulated in tumour tissue.
When investigating the dependence of overall survival and disease-free survival on relative gene expression, in the overall group of patients, we found correlation of VSNL1 expression with OS (significantly longer OS was observed among patients with VSNL1 expression level in tumour tissues above the median value; Cox-Mantel p=0.033 and log rank p=0.032) (Figure 2A).
In the adjuvant group, patients with SLC26A2 expression above the tumour tissue median had a longer OS than those with an expression level below the median (Cox-Mantel p=0.036) (Figure 2B). In the case of DFI, patients with VSNL1 expression in the healthy tissue below the median had a longer DFI than patients with higher expression (Cox-Mantel p=0.022, log-rank p=0.020) (Figure 2C). The opposite trend was observed for SLC26A2 (Cox-Mantel p=0.012, Wilcoxon p=0.014 and log-rank p=0.011) (Figure 2D).
Then we analyzed the OS and DFI of the patients with respect to the difference in candidate gene expression between healthy and tumour tissues. In the overall group of patients, we found that the greater the down-regulation of CLDN23 expression in tumour, the worst the outcome for the patient in terms of shorter OS (Wilcoxon p=0.045) (Figure 2E). In the adjuvant group, a smaller decrease of SLC26A2 and ACSL5 expression in tumour led to a longer DFI (Wilcoxon p=0.046, Cox-Mantel p=0.045 and log-rank p=0.041 for SLC26A2, and Wilcoxon p=0.040 for ACSL5) (Figure 2F and 2G) as did a greater increase of LGR5 expression (Wilcoxon p=0.046, Cox-Mantel p=0.028 and log-rank p=0.026) (Figure 2H).
Impact of tumour location on candidate gene expression. Patients were divided into two groups according to the location of the primary CRC and gene expression was analysed using the Mann-Whitney U-test. The groups did not exhibit any significant difference in candidate gene expression in tumour tissue, but we found differential expression of MAPK1 (p=0.032), LGR5 (p=0.003), MIER3 (p=0.026) and CTHRC1 (p=0.042) in healthy mucosa. All of these genes were expressed more highly in the group of patients with tumours localised in the colon. Regarding the difference in expression, we found a significant difference in VCAN expression change between patients with colon localisation (smaller increase) and those with rectosigmoid/rectum-localised tumours (greater increase) (p=0.031).
Discussion
The relationship between the change of gene expression and potential functional effects is not usually straightforward. In the following paragraphs, we briefly summarise present knowledge about the individual genes analyzed here and discuss our results with respect to current literature, focusing on possible explanations of the role of these expression changes on the clinical behaviour of colorectal tumours. Because of the heterogeneity of the selected genes, we discuss individual genes separately.
VSNL1. This gene encodes neuronal calcium sensor protein which can modulate the activity of adenylate cyclase (18) and has been described to be a prognostic marker in Alzheimer's disease (19, 20). VSNL1 is also considered to be a potentiator of invasiveness and inhibitor of proliferation of neuroblastoma cells (21), a tumour suppressor in non-small cell lung carcinomas (22), and an inhibitor of epithelial-to-mesenchymal transition in squamous carcinoma cells (23).
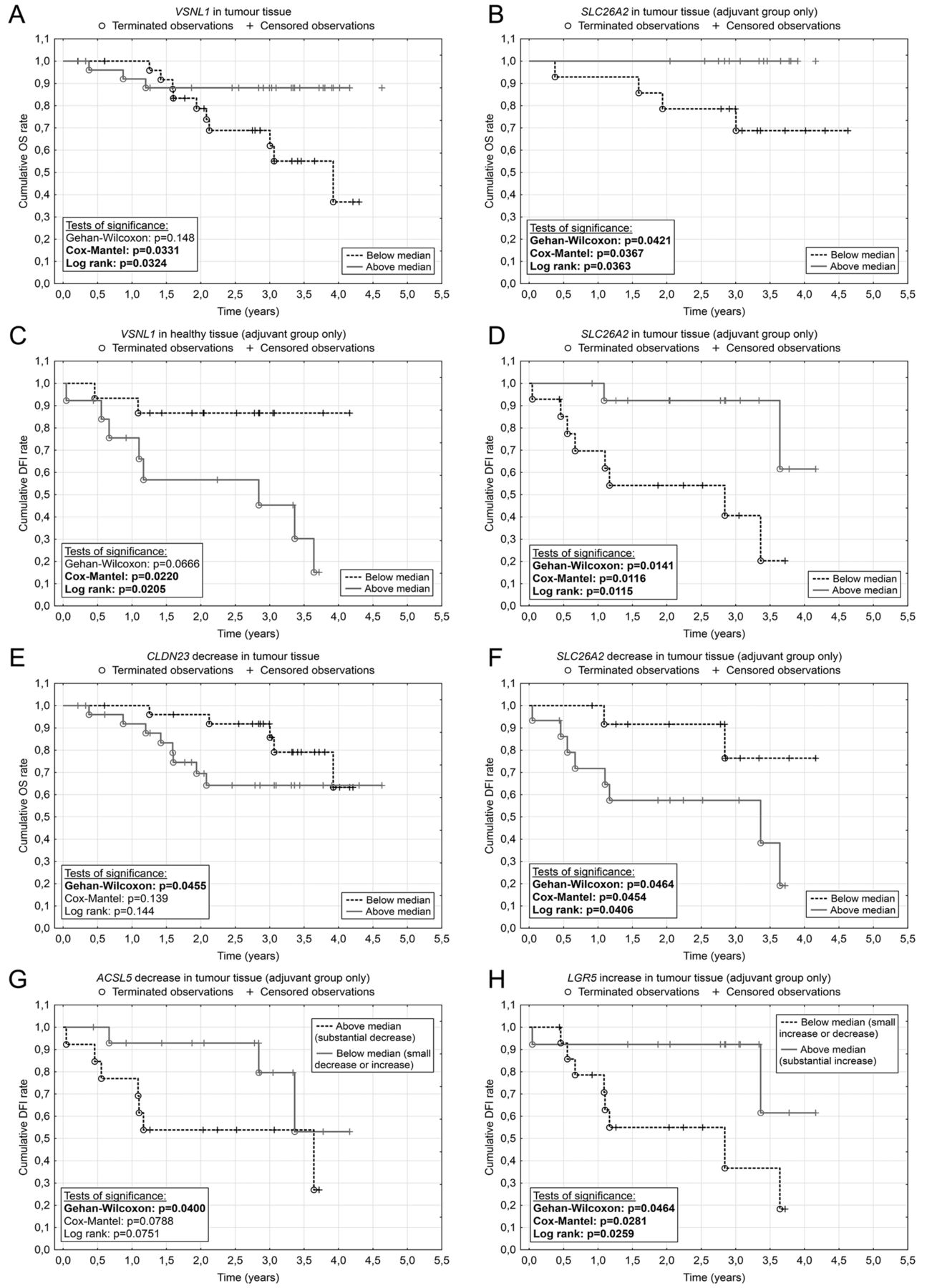
Kaplan-Meier curves of statistically significant associations between gene expression and disease-free survival (DFI)/overall survival (OS). Relation of the gene expression in the tumour (TT) and healthy (HT) tissue to the DFI and OS (A-D). Relation of gene expression change to the DFI and OS (E–H). Patients were divided by the median of the expression change. In the case of ACSL5, one group consisted of patients with substantial decrease of ACSL5 expression and the second group consisted of patients with slight decrease or increase of ACSL5 expression. Similarly, for LGR5, we grouped patients with small increase or decrease of expression and compared them with patients with a substantial increase in LGR5 mRNA. A: Visinin-like 1 (VSNL1); B: Solute carrier family 26 (sulfate transporter), member 2 (SLC26A2); C: Visinin-like 1 (VSNL1); D: Solute carrier family 26 (sulfate transporter), member 2 (SLC26A2); E: Claudin 23 (CLDN23); F: Solute carrier family 26 (sulfate transporter), member 2 (SLC26A2); G: Acyl-CoA synthetase long-chain family member 5 (ACSL5); H: Leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5).
In colon cancer, VSNL1 was found to be down-regulated after selenomethionine-induced growth arrest of the colon cancer cell line HCT116 (24) and it was described to be a putative marker for identification of circulating tumour cells in peripheral blood (25). We did not observe the correlation of VSNL1 overexpression with lymph node metastasis that was described earlier (26), but we confirmed overexpression of VSNL1 in tumour tissue compared to healthy mucosa, and, more importantly, we found a relation between higher expression of VSNL1 in tumour tissue and longer survival of patients, hence our results should support the possible function of VSNL1 as tumour suppressor as reported for other types of tumour (22, 27). The opposite effect of VSNL1 expression in healthy tissue on DFI is the subject of further investigation.
VCAN. Versican protein, encoded by the VCAN gene, is one of the major components of the extracellular matrix (ECM) with various functions in the regulation of cell migration (28), proliferation (29) and cell adhesion (30). VCAN was shown to play a role in many types of cancer, including gastric (28), pancreatic (31) and colorectal (32).
We found an up-regulation of VCAN mRNA in tumour, which is in agreement with previously described changes of expression on the protein level, which was accompanied by altered post-translation modifications (32, 33). The level of up-regulation found here was different depending on the tumour location (colon versus rectosigmoid/rectum).
LGR5. Leucine-rich repeat-containing G-protein-coupled receptor 5 protein is a wingless-type MMTV integration site family (WNT) signaling target and a marker of intestinal (34) and liver stem cells (35). Expression of LGR5 is increased in intestinal tumours, and can be found in cancer cells with stem cell properties (36, 37).
In our study, we observed an up-regulation of LGR5 expression in tumour tissue, and a greater increase of expression in tumour tissue compared to healthy tissue correlated significantly with a longer DFI in adjuvantly treated patients, which is contradictory to previously published data, where overexpression in malignant tissue was shown to have adverse effects on disease outcome in CRC and other types of tumours (38, 39, 40). This finding needs to be further evaluated.
CTHRC1. CTHRC1 exhibited the highest up-regulation in tumour tissue among the genes we analyzed. CTHRC1 gene encodes a protein involved in vascular remodeling (41) and an expression change has been described in many types of solid tumours (42), including CRC (43). Recently, the expression status of CTHRC1 was identified as being a predictor of poor prognosis in CRC patients (44). We confirmed the expression change, but we were not able to significantly prove the prognostic role of CTHRC1 expression at a p-value below 0.05. Nevertheless, we identified a trend for shorter survival of patients with higher CTHRC1 expression levels in tumour tissues.
SLC26A2. In mammals, there are 11 genes of the SLC26 family, which function as anion exchangers or channels. The main role of SLC26A2 is to transport SO42− anions and its altered function was related to several types of chondrodysplasias (45).
Lower expresion of SLC26A2 was related to enhanced proliferation of colon cancer cells in vitro (46). A decrease of expression was also observed in bioptic samples (47), and we detected a severe down-regulation of SLC26A2 in tumour tissue. Based on these data, we can conclude that SLC26A2 down-regulation is also important for tumour propagation in vivo. The expression level of SLC26A2 was associated with the decrease of OS and DFI in the adjuvantly treated group of patients. A probable effect of down-regulation of SLC26A2 is disorganisation of the ECM by improper sulfation of ECM proteins, therefore helping tumour cells to migrate through the tissue.
CLDN23. This gene belongs to the claudin family of 24 genes encoding proteins with four transmembrane domains involved in formation of tight junctions among adjacent cells (48). CLDN23 itself was described to be down-regulated in intestinal-type gastric cancer (49), but there are no studies focused on this gene in a larger set of patients with colon cancer.
In our set of patients, CLDN23 was down-regulated in tumour tissue and we found that the level of down-regulation correlates with the OS: a smaller difference of CLDN23 expression between tumour and normal tissue was associated with a better prognosis. This effect can be explained by the fact that for the migration of tumour cells, it is neccessary to break the bonds between adjacent cells and strong down-regulation of tight junction proteins, including CLDN23, is one such crucial step in this proscess. Therefore, patients with very low CLDN23 expression would be more prone to progression of CRC.
ACSL5. This gene encodes an enzyme implicated in lipid biosynthesis and fatty acid degradation. ACSL5 down-regulation was associated with small intestine carcinoma (50), but ACSL5 was found to be up-regulated in CRC by microarray analysis (51). In gliomas, it can be considered as a cancer survival factor (52). The function of ACSL5 is probably dependent on the particular splice variant, since the full-length ACSL5 is pro-apoptotic (53). Our analysis found ACSL5 to be down-regulated in tumour tissue, and we also identified that the greater the decrease, the shorter the DFI observed in the adjuvantly-treated group of patients, assuming a protective function for ACSL5.
DSTN, MIER3 and MAPK1. These three genes were found to be down-regulated in tumour tissues, but in our set of samples, we did not identify any correlation with clinical characteristics.
The product of the DSTN gene is the actin-binding protein destrin, also known as actin-depolymerizing factor, which has an important function in regulation of actin dynamics (54). Regarding the role of DSTN in cancer development and progression, there are several pieces of information about its role in membrane androgen receptor-induced apoptosis of prostate cancer cells (55), and its positive regulation of the migration of neuroblastoma cells (56); the down-regulation of DSTN also blocks migration and invasive capacity of gastric cancer (57). We found a slight down-regulation in tumour tissue, but this change did not correlate with any clinical parameter.
MIER3 is a practically unresearched gene, but in 2012, it was shown that it probably plays a role in tumourigenesis because it was described as a candidate breast cancer susceptibility gene (58) and also a highly mutated gene in hypermutated colorectal tumours (59). Our data suggest down-regulation in CRC, but without strong correlations to clinical data.
Mitogen-activated protein kinase 1 is the final kinase in the MAPK pathway, which has a pleiotropic role like regulation of proliferation, differentiation and gene transcription (60). In CRC, down-regulation of MAPK1 was shown in a previous study (61), as well as in our set of samples.
CAPN10 and SAMD3. The function of sterile alpha motif domain-containing 3 is unknown; this gene was selected because of its possible up-regulation during CRC development (13). This gene is interesting regarding its observed interaction with Fanconi anemia, complementation group G (FANCG) (62), which plays a role in the maintanence of genomic integrity (63), but we did not observe any change of its expression. This was also the case with CAPN10, a member of the calcium activated cystein proteases (64). Particular single nucleotide polymorphisms in the CAPN10 gene were shown to be associated with type 2 diabetes mellitus (65), and another was also associated with CRC susceptibility (66).
Conclusion
We have described significant expression changes of 10 studied genes in the CRC tissue compared to healthy mucosa. We also identified associations between different gene expressions and overall and disease-free survival, which could provide with useful information for disease prognosis and patient follow-up. We identified several putative positive prognostic factors, such as ACSL5 and CLDN23, in tumour tissues. Our results will serve for the development of a disease risk scoring system based on the expression of these selected genes. This system will be validated and evaluated on a new group of patients with CRC. The most promising genes (mainly up-regulation of SLC26A2 and down-regulation of CLDN23 and ACSL5) will be further studied individualy on a larger set of patients.
Acknowledgements
The publication of this article was supported by grants IGA MZ CR 12025 and 14329, grant GAUK 12536, project CZ.1.05/2.1.00/03.0076 from the European Regional Development Fund and the Charles University Research Fund (project number P36). Study was approved by Institutional Review Board and Ethical Comitee of Teaching Hospital and Medical Faculty in Pilsen number 421/2012.
- Received August 25, 2013.
- Revision received September 25, 2013.
- Accepted September 27, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Colon Cancer Progression Is Reflected to Monotonic Differentiation in Gene Expression and Pathway Deregulation Facilitating Stage-specific Drug Repurposing
- Altered Expression of MBNL Family of Alternative Splicing Factors in Colorectal Cancer
- Collagen Triple Helix Repeat Containing 1 Expression in Osteosarcoma: A New Predictor of Prognosis