


Abstract
Aim: To evaluate the effects of HSP90 blockade by EC154 on the oncogenic receptor tyrosine kinase macrophage-stimulating 1 receptor (MST1R) in gastric and pancreatic cancer. Materials and Methods: Impact of EC154 on signaling pathways was investigated by western blotting. Cancer cell migration was evaluated in Boyden chambers. Transcriptional regulation of MST1R was examined by using promoter-luciferase reporter constructs. Effects on MST1R expression, and tumor growth were investigated in in vivo tumor models. Results: MST1R was expressed by cancer cells without evidence of MST1R mutations. EC154 led to an effective inhibition of cancer cell growth, down-regulated MST1R, diminished its promoter activity, and disrupted oncogenic macrophage-stimulating protein 1 (MSP1) signaling. Moreover, pro-migratory activities of cancer cells were dramatically inhibited. In vivo, treatment with EC154 significantly reduced tumor growth, while MST1R expression was down-regulated. Conclusion: Wild-type MST1R is an HSP90 client protein that can be targeted in gastrointestinal cancer using HSP90 inhibitors.
Receptor tyrosine kinases (RTKs) are known to be involved in critical processes of cancer progression, including cancer cell proliferation, invasion, and metastasis. Within the last decade, numerous molecular agents have been developed to target certain RTKs in terms of inhibiting their activity. In this context, the RTK macrophage-stimulating 1 receptor (MST1R), also known as recepteur d'origine nantais (RON), has recently gained great interest as a target for therapy against various cancer entities, including pancreatic cancer (1-3). MST1R is a 180-kDa protein that belongs to the mesenchymal epithelial transition factor (MET)-proto-oncogene family and is involved in multiple signaling cascades that mediate adhesion, cell motility, proliferation, and apoptosis (4). Several oncogenic signaling pathways are located downstream of MST1R, including RAS/mitogen-activated protein kinase (MAPK), phosphatidyl inositol-3 kinase (PI-3K)/AKT, and focal adhesion kinase (FAK) pathways (5). Importantly, MST1R and its ligand, macrophage-stimulating protein (MSP), have been implicated in cancer progression and metastasis. Overexpression of MST1R in colon, breast, and ovarian cancer cell lines enhances migratory and invasive properties of cancer cells (6-8). Regarding pancreatic cancer, the oncogenic role of MST1R expression has been clearly demonstrated in human pancreatic intraepithelial neoplasia, and in primary human and mouse metastatic cell lines (9). Moreover, activation of MST1R induces molecular and cellular alterations that are consistent with the process of epithelial mesenchymal transition (EMT), suggesting that MST1R signaling may contribute to pancreatic carcinogenesis (10). Furthermore, overexpression of MST1R has been reported for patients with gastric cancer (11). Thus, targeting MST1R might be an effective therapeutic strategy for the treatment of certain cancer entities, including pancreatic and gastric cancer.
Recently, the molecular chaperone heat-shock protein 90 (HSP90) has been identified as being a crucial component for the stability and function of a wide variety of oncogenic molecules and signaling intermediates, including epidermalgrowth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER-2), c-MET, mutated p53, AKT, ERK, and hypoxia-inducible factor (HIF)-1α (12-14). Due to its central role in oncogenic signaling, HSP90 provides an attractive target for the treatment of cancer. Inhibition of HSP90 function results in a simultaneous disruption of numerous signal transduction pathways which are pivotal to tumor progression and cell survival. Using experimental models of gastric and pancreatic cancer, we were able to demonstrate that these tumor entities harbor multiple oncogenic HSP90 client proteins which can effectively be controlled by HSP90 inhibition, thus leading to significant growth reduction in vivo (15, 16). Interestingly, evidence exists that MST1R could be a novel HSP90 client since studies have shown that mutated MST1R is susceptible to HSP90 inhibitor-mediated degradation (2, 17-19). Furthermore, such MST1R mutations (M1254T) have artificially been introduced into cancer cells in these reported studies (2, 18, 19).
In the present study, we hypothesized that oncogenic MSP/MST1R signaling can be targeted by HSP90 inhibitors and we investigated the effects of a novel synthetic HSP90 inhibitor (EC154) in human pancreatic and gastric cancer cells both in vitro and in vivo.
Materials and Methods
Cells and reagents. The human pancreatic cancer cell line HPAF-II and the human gastric cancer cell line AGS were obtained from the American Type Culture Collection (Manassas, VA, USA), and the metastatic L3.6pl cell line was kindly provided by Dr. I.J. Fidler (The University of Texas MD Anderson Cancer Center, Houston, TX, USA). The human gastric cancer cell line TMK-1 was obtained from Dr. Eiichi Tahara (University of Hiroshima, Hiroshima, Japan). Cells were cultured in Dulbecco's modified eagle medium (DMEM; Life Technologies, Darmstadt, Germany) supplemented with 10% or 15% fetal bovine serum (FBS; Life Technologies, Darmstadt, Germany) and maintained in 5% CO2 at 37°C, as described elsewhere (20, 21). All in vitro experiments were performed at 60% to 70% cell confluence. For in vivo experiments, trypsinized cells were resuspended in Hank's balanced salt solution (HBSS). The synthetic HSP90 inhibitor EC154 was kindly provided by Biogen Idec (San Diego, CA, USA) and represents a next-generation HSP90 inhibitor with both i.v. and oral bioactivity. The HSP90-binding activity of EC154 was confirmed, as described below. Recombinant human MSP1 was purchased from R&D Systems (Minneapolis, MN, USA).
HSP90 binding assay for EC154. Recombinant human HSP90 (0.8 nM; Biomol GmbH, Hamburg, Germany) and reduced Fluorescein isothiocyanate (FITC)-geldanamycin (2 nM; Biomol GmbH, Hamburg, Germany), reduced with Tris(2-carboxyethyl)phosphine (TCEP; Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) for 3 h at room temperature, were incubated in a 96-well microplate at room temperature for 3 h in assay buffer containing 20 mM Hepes, (pH 7.4), 50 mM KCl, 5 mM MgCl2, 20 mM Na2MoO4, 2 mM DTT, and 0.1 mg/ml bovine gamma globulin. Following the pre-incubation, competitor compound in 100% Dimethyl sulfoxide (DMSO; Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) was added to final concentrations between 0.04 nM and 10 μM (final volume 100 μl, 2% DMSO). The reaction was incubated for 16 h at room temperature with gentle shaking. Fluorescence polarization in mP was measured in an EnVision plate reader (Perkin Elmer, Waltham, MA, USA) using 485 nm excitation and 535 nm emission wavelengths. Maximum signal and background controls contained no compound and no protein, respectively. The data were fit to a 4-parameter curve using GraphPad Prism (GraphPad Software, La Jolla, CA, USA), and IC50 values were generated. The inhibition constant (Ki) values were calculated using a modified Cheng-Prusoff equation: Ki=[I]50/([L]50/Kd+[P]0/Kd+1). Where [I]50 is the free inhibitor concentration at 50% inhibition; [L]50 is the free tracer concentration at 50% inhibition; Kd is the dissociation constant for interaction of the protein and tracer (HSP90 Kd=1 nM); and [P]0 is the initial protein concentration. EC154: HSP90: average IC50: 5.2±1.9 nM (n=8); average Ki: 1.6±0.6 nM.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. To evaluate the antiproliferative and cytotoxic potential of EC154, gastric and pancreatic cancer cells were seeded into 96-well plates (1 × 103 cells per well) and exposed to different concentrations of EC154 (50-700 nM) for the indicated times at 37°C. We used the MTT assay 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay to assess cell viability, as previously described (15).
Western blot analysis of signaling intermediates. To determine the effects of EC154 on signaling intermediates, western blot analysis was performed. Experiments were performed in triplicates at a cell density of 60% to 70% confluency. Unless otherwise indicated, cells were incubated with EC154 (200 nmol/l) for 24 h before stimulation with MSP1 (100 ng/ml). Whole cell lysates were prepared as described elsewhere (16). Protein samples (75 μg) were subjected to western blotting using a denaturing 10% sodium dodecyl sulfate polyacrylamide gel. Membranes were sequentially probed with antibodies specific for phospho-MEK, MEK, phospho-AKTSer473, AKT, phospho-ERKThr202/Tyr204, ERK, phospho-signal transducer and activator of transcription 3 (STAT3)Tyr705, STAT3, phospho-MST1R and MST1R (all from Cell Signaling Technologies, Beverly, MA, USA), insulin-like growth factor 1 receptor β (IGF1Rβ) and β-actin (Santa Cruz Biotechnologies, Santa Cruz, CA, USA). Antibodies were detected by enhanced chemiluminescence (Amersham Bioscience, Piscataway, NJ, USA). Western blot analyses of tumor tissue samples were carried out likewise after tissue lysis using an extraction buffer, as described elsewhere (15).
MST1R promoter cloning. The human MST1R promoter region was amplified by polymerase chain reaction (PCR) and cloned into the pGL3-Basic luciferase plasmid (Promega GmbH, Mannheim, Germany) in two-rounds of amplification from total human genomic DNA, as described elsewhere (22, 23). In the first-round, the amplification profile was one cycle of 94°C for 5 min; 35 cycles of 94°C for 40 s, 70°C for 40 s and 72°C for 3 min; one cycle of 72°C for 10 min. The PCR fragment was cloned into PCRII vector (Life Technologies, Darmstadt, Germany) following the manufacturer's instructions. The fragment was a 3000 bp-length construct. The second-round amplification used 1 ng of PCRII vector cloned first-round product, amplification profile was one cycle of 94°C for 5 min; 35 cycles of 98°C for 20 s, 55°C for 30 s and 72°C for 50 s; one cycle of 72°C for 10 min. The fragment was sub-cloned into pGL3-Basic, following restriction digestion with BglII and KpnI, using standard DNA ligation techniques producing the 456 bp-length construct. The primers used for PCR were as follows: sense; 5’-GGTACCTAGCTGACCTCCCCCACACACCTTGACTGC-3’; antisense; 5’-AGATCTACTGGGCCAAATTTAAGCAGCGGTCCCGAC-3’. The nucleotide sequences of construct were confirmed by DNA sequencing (22, 23).
Transient transfection of MST1R and early growth response 1 (EGR1)-reporter and luciferase assay. The transcriptional regulation of MST1R and EGR1 was examined by the transient transfection of a EGR1-, and MST1R-promoter-luciferase reporter construct, respectively (24). The plasmid EGR1-promoter was kindly provided by Dr. Y.H. Lee (Konkuk University, Seoul, Korea). AGS cells were seeded in 6-well plates and transfected with 1 μg of the EGR1-promoter construct using Lipofectamine 2000 (Life Technologies, Darmstadt, Germany) according to the manufacturer's instructions. To monitor transfection efficiency, 50 ng of the pRL-null plasmid encoding Renilla luciferase were included in all samples. At 24 h post-transfection, the levels of firefly and Renilla luciferase activity were measured sequentially from a single sample using the Dual-Glo Luciferase Assay System (Promega GmbH, Mannheim, Germany). The firefly luciferase activity was normalized to that of Renilla, and the relative amount of luciferase activity in the untreated cells was designated as 1. The luminescence was measured with a dual luminometer.
The effects of HSP90 inhibition on phorbol myristic acetate (PMA)-induced EGR1 activity were investigated by pretreating transfected cells with EC154 for 1 h at the indicated doses with subsequent treatment with PMA for 6 h. Afterwards, cells were lysed for luciferase assays.
Migration assays. To determine the effects of EC154 (200 nmol/l) on cancer cell motility in vitro, migration assays were performed using modified Boyden chambers, as described elsewhere (25). Briefly, 1 × 105 cells were resuspended in 1% FCS-DMEM and seeded into inserts with 8-mm filter pores, and 10% FCS-DMEM, with or without MSP1 (100 ng/ml) which served as a chemoattractant. After 24 h and 48 h, cells were fixed and migrated cells were stained (Diff-Quick reagent; Dade Behring, Newark, NJ, USA). Cells were counted in four fields, and average numbers were calculated.
Animal models. Eight-week-old male athymic nude mice (BALB/cnu/nu; Charles River, Sulzfeld, Germany) were used for experiments, as approved by the Institutional Animal Care and Use Committee of the University of Regensburg and the regional authorities. In addition, experiments were conducted according to Guidelines for the Welfare of Animals in Experimental Neoplasia published by The United Kingdom Coordinating Committee on Cancer Research (26). The effects of HSP90 inhibition on the growth of human gastric (TMK-1) and pancreatic cancer cells (L3.6pl) were investigated in a subcutaneous xenograft tumor model. Cancer cells (1 × 106) were injected into the subcutis (right flank) of nude mice. Mice were randomized (n=8 per group) and assigned to treatment groups. Intraperitoneal injections of EC154 (25 mg/kg, or 50 mg/kg, twice per week) were started on day 5 when tumors became palpable. Tumor diameters were measured every other day and tumor volumes were calculated (width2 × length × 0.5). When the experiment was terminated on day 13 after tumor cell injection, tumors were excised, weighed and prepared for western blot analyses.
The effects of EC154 were additionally evaluated in an orthotopic pancreatic cancer model, as described previously (20). In brief, 5 × 105 human pancreatic cancer cells (L3.6pl) were injected into the pancreas of mice. Tumors were allowed to grow for 7 days after implantation, before treatment was initiated. Mice were randomized into groups (n=10 per group) receiving either vehicle (saline) or EC154 (25 mg/kg, twice per week) by i.p. injections. On day 28 after tumor cell inoculation, mice were sacrificed and excised tumors were measured, weighed and prepared for western blot analyses. For immunohistochemical analyses, tumors were either paraffin embedded or placed in optimum cutting temperature (OCT) solution.
Immunohistochemical analysis of tumor cell proliferation. Multiple cryosections were obtained from tumors for immunohistochemical analyses. To measure proliferating tumor cells, mice received 1.0 mg per mouse i.p. injections of bromodeoxyuridine (BrdUrd; Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) 2 h before the termination of animal studies. A BrdUrd detection kit (Becton Dickinson GmbH, Heidelberg, Germany) was used to visualize BrdUrd uptake of cells in sections of tumors. Briefly, sections were incubated with anti-BrdUrd antibody solution followed by streptavidin-conjugated horseradish peroxidase-linked goat anti-mouse IgG2. Antibody binding was visualized by incubating slides in diaminobenzidine with hematoxylin counterstaining. BrdUrd-positive tumor cells were counted in four fields per tumor section at ×20 magnification and averages were calculated (27).
Statistical analyses. Statistical analyses were carried out using SigmaStat (version 3.0; Systat Software GmbH, Erkrath, Germany). Results of in vivo experiments were analyzed for outliers using the Grubb's test (http://www.graphpad.com). Tumor-associated variables in in vivo experiments were tested for statistical significance using the Mann-Whitney U-test. The two-sided Student's t-test was applied for analysis of in vitro data. All results are expressed as the mean±SE.
Results
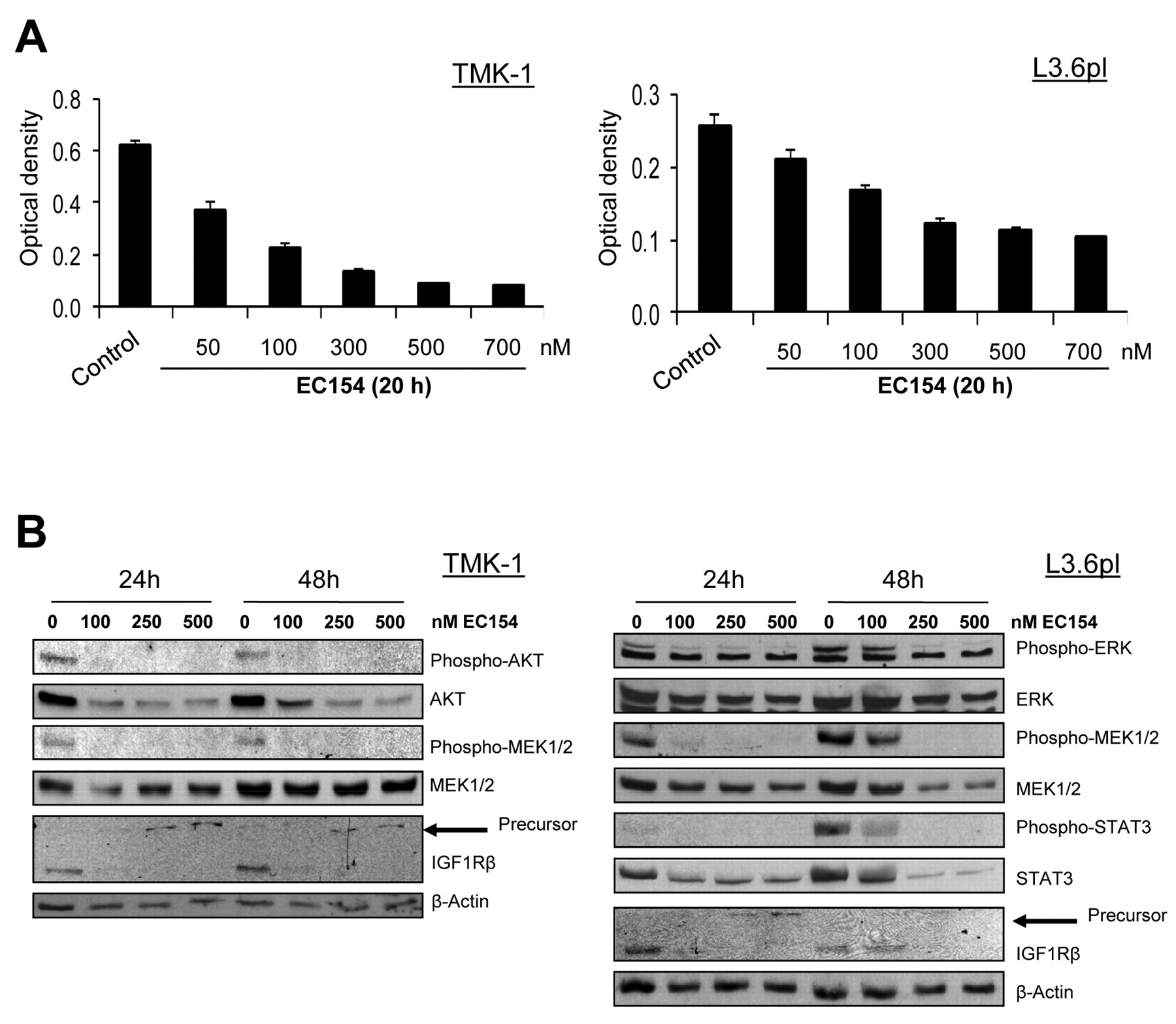
Effects of HSP90 inhibition on pancreatic and gastric cancer cells in vitro. The cytotoxic and antiproliferative effects of HSP90 inhibition with EC154 were first determined by MTT analysis, showing that EC154 leads to an effective dose-dependent inhibition of both gastric and pancreatic cancer cell growth (Figure 1A). To identify the intracellular signaling pathways that are affected by this inhibitor, we next investigated whether EC154 treatment leads to alterations in constitutively activated pathways that have been implicated in the regulation of gastric and pancreatic cancer growth. Western blot analyses showed that EC154 (100-500 nM/l) strongly reduces the constitutive phosphorylation of AKT, and MEK in gastric and of ERK1/2, MEK and STAT3 in pancreatic cancer cells (Figure 1B). In addition, IGF1R is substantially down-regulated by blocking HSP90 with EC154, an effect which was paralleled by up-regulation of the IGF1Rβ precursor protein, as observed in our previous studies in pancreatic cancer using geldanamycin derivates (16). We conclude that EC154 effectively interferes with important pro-tumorigenic signaling cascades in gastric and pancreatic cancer cells.

Effects of HSP90 inhibition on tumor cells. The effects of the HSP90 inhibitor EC154 on gastric (TMK-1) and pancreatic (L3.6pl) cancer cells were determined in vitro. A: Growth-inhibitory effects of EC154 were determined by MTT analyses. EC154 potently reduced growth of both gastric and pancreatic cancer cells. B: Effects of EC154 on the activation of signaling pathways were investigated by western blotting. EC154 led to a robust inhibition of AKT, MEK, ERK1/2, and signal transducer and activator of transcription 3 (STAT3)-activation in the cell lines. Importantly, insulin-like growth factor 1 receptor (IGF1R) was down-regulated by HSP90 inhibition.
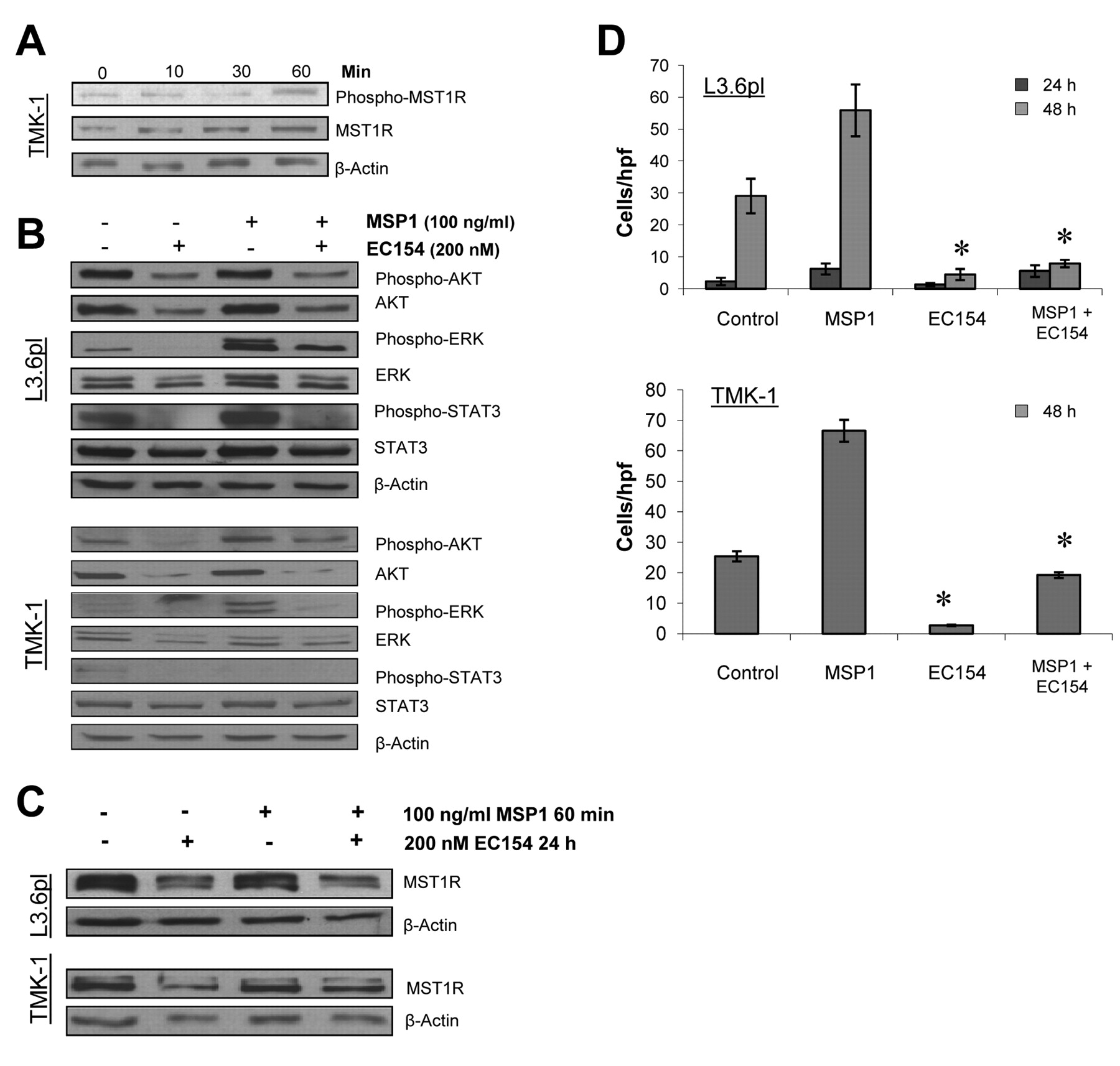
Effects of HSP90 inhibition on the MST1R system. Stimulation of gastric (TMK-1) and pancreatic cancer cells (L3.6pl) with MSP1 led to an activation of the MST1R system in cancer cells (Figure 2A). Furthermore, treatment with EC154 substantially disrupted MST1R signaling in terms of diminishing both downstream phosphorylation and total protein levels of the substrates ERK1/2, AKT, and STAT3 in cancer cells (Figure 2B). Interestingly, the inhibition of HSP90 using EC154 also elicited a direct effect on MST1R function in terms of down-regulating MST1R expression per se (Figure 2C). Since mutant MST1R only has so far been associated with a high susceptibility to HSP90 inhibition, sequencing of cancer cell lines was performed to detect MST1R mutations. None of the investigated cell lines harbored mutations of MST1R, suggesting that wild-type MST1R represents a novel HSP90 client protein (data not shown). As a functional consequence of multiple pathway and receptor inhibition, EC154 significantly inhibited MSP1-induced cancer cell migration of gastric and pancreatic cancer cells (Figure 2D).
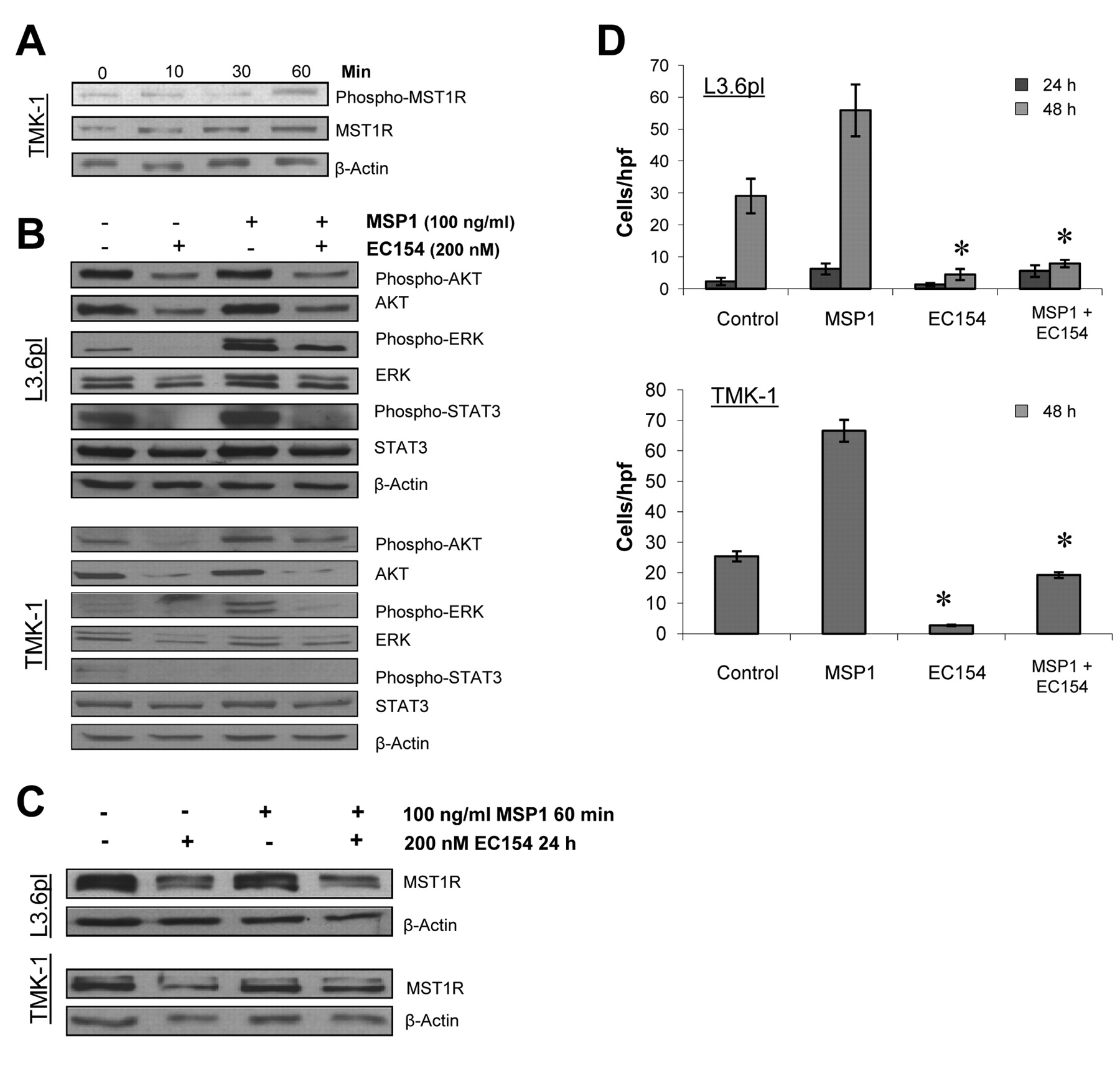
Effect of HSP90 inhibition on macrophage-stimulating protein 1 (MSP1) signaling and macrophage-stimulating 1 receptor (MSTR1) expression. A: MSP1-mediated MST1R activation and MST1R expression were investigated by western blotting. Cancer cells were stimulated with MSP1 at 100 ng/ml for the indicated times. Results are presented for TMK-I gastric cancer cells. B: Blockade of HSP90 substantially disrupted MSP1-mediated activation of AKT, ERK1/2, and signal transducer and activator of transcription 3 (STAT3) in pancreatic and gastric cancer cells. C: Inhibition of HSP90 by EC154 led to a down-regulation of MST1R in gastric (TMK-1) and pancreatic (L3.6pl) cancer cells. D: Changes in cell migration were investigated in uncoated Boyden chambers. EC154 (200 nM) significantly reduced MSP1-mediated (100 ng/ml) migration of human pancreatic and gastric cancer cells (L3.6pl, TMK-1) (*p<0.05). Bars=mean±SEM.
The functional effects on MST1R were further investigated by validating the results of protein analysis by PCR. Since phorbol myristic acetate (PMA) is a strong inducer of MST1R via PKC signaling, this compound was used for measuring HSP90 inhibitor-modulated MST1R promoter activity. PMA strongly induced both MST1R mRNA and protein expression in gastric cancer cells, an effect which was diminished by blocking HSP90 with EC154 (Figure 3A, and B). Moreover, PMA-induced MST1R promoter activity can be reduced by EC154 treatment (Figure 3C) and this reduction in MST1R promoter activity subsequently diminishes the expression of the M5T1R down-stream element EGR1 (Figure 3D). Yet again, EGR1 promoter activity in part appears to be susceptible to HSP90 inhibition (Figure 3E). We conclude from these experiments that blocking HSP90 leads to functional inhibition of MST1R.

Effect of HSP90 inhibition on MST1R activity. Phorbol myristic acetate (PMA), a tumor promoter by activating PKC, was used for up-regulating MST1R in human gastric carcinoma cells (AGS). The cells were pretreated with EC154 at the indicated concentration and subsequently stimulated for 1 h with PMA. A: After 4 h, RT-PCR showed the effects of HSP90 inhibition on PMA-induced MST1R expression. Blocking HSP90 with EC154 reduced MST1R expression and PMA-mediated induction. B: Similar results were obtained at the protein level, showing that HSP90 inhibition diminishes MST1R expression. C: The MST1R promoter activity luciferase assay shows that the HSP90 inhibitor EC154 diminishes PMA tumor promoter-induced MST1R expression. D: The transcription factor early growth response 1 (EGR1) represents a down-stream target of MST1R. PMA was used for up-regulating EGR1 in human gastric carcinoma cells (AGS). Blocking HSP90 reduced inducible EGR1 protein levels. E: Moreover, as determined by EGR1 promoter luciferase assay, EC154 diminished tumor promoter-induced EGR1 expression.

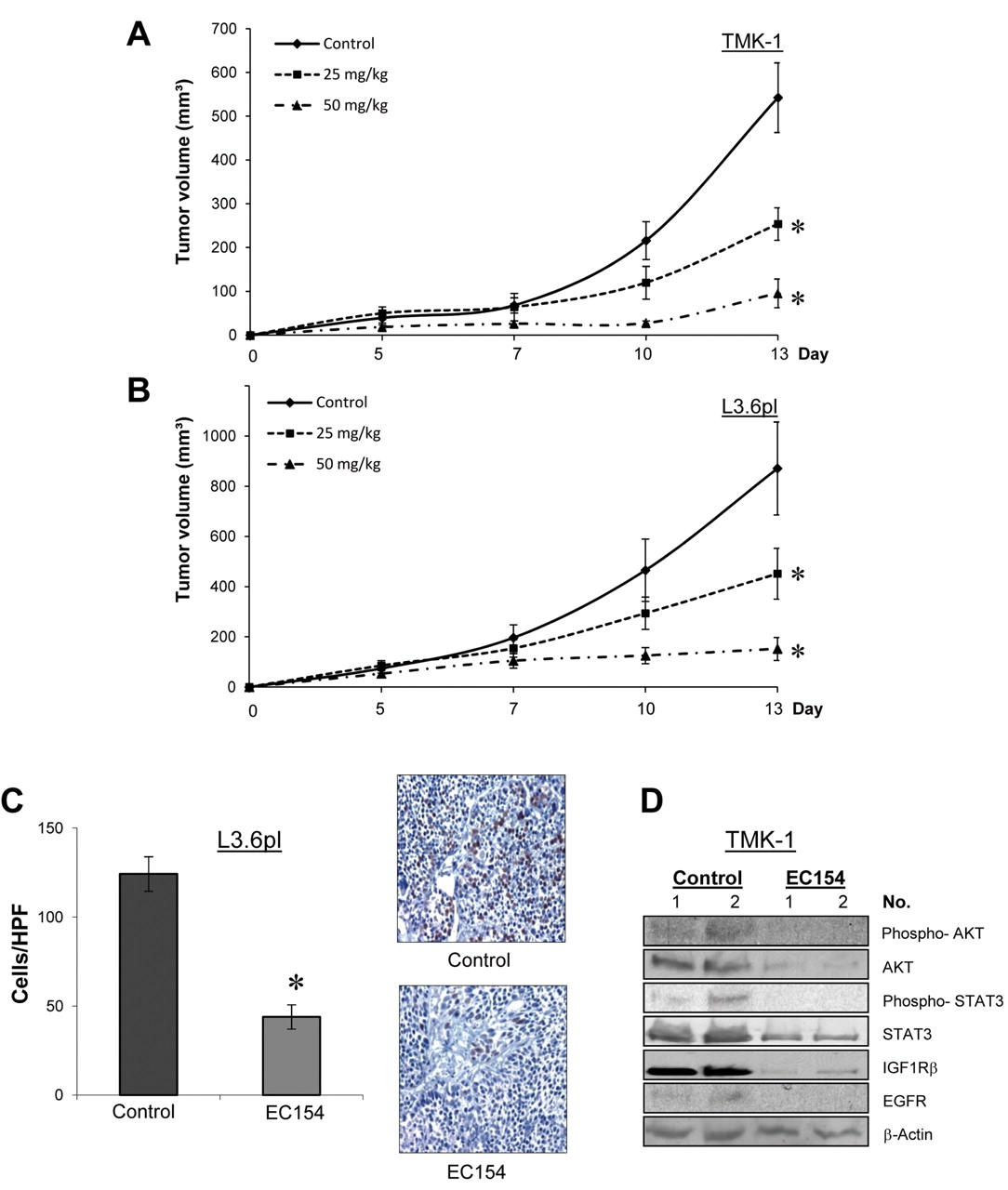
Effect of EC154 on subcutaneous tumor growth. The effects of EC154 (25 mg/kg, or 50 mg/kg; twice per week, i.p.) on tumor growth were investigated in subcutaneous models (n=8/group). EC154 significantly reduced the growth rates of both gastric (A) and pancreatic (B) cancer cells in vivo (*p<0.05). C: Treatment with EC154 significantly reduced the number of proliferating (BrdU-positive) tumor cells in tissue sections, as determined in pancreatic tumors (*p<0.01). D: Western blot analyses of gastric tumor tissues (EC154; 50 mg/kg) show that EC154 leads to inhibition of AKT- and signal transducer and activator of transcription 3 (STAT3)-activation. The expression of insulin-like growth factor 1 receptor β (IGF1Rβ), as well as epidermal growth factor receptor (EGFR) is effectively reduced by EC154 treatment. Bars=mean±SEM.

Effect of EC154 on orthotopic pancreatic tumor growth. The effects of EC154 therapy were investigated in an orthotopic pancreatic cancer model (n=10/group). Treatment started on day 7 (25 mg/kg, twice per week, i.p.) when tumors were established. EC154 significantly reduced pancreatic tumor growth, as reflected by final tumor volumes (A) and tumor weights (B) (*p<0.05 for both). C: Western blot analyses of pancreatic cancer tissues (EC154; 25 mg/kg) show that EC154 leads to a marked reduction in MST1R expression in vivo. Bars=mean±SEM.
Effects of EC154 on tumor growth and MST1R expression in vivo. The effects of HSP90 inhibition on cancer growth in vivo were first determined in subcutaneous xenograft models of pancreatic (L3.6pl) and gastric (TMK-I) cancer. Treatment with EC154 dose-dependently inhibited the growth of both gastric and pancreatic tumors (Figure 4A, and B) (p<0.05). This potent growth-inhibitory effect was also mirrored by the weights of the excised tumors, which were significantly reduced in EC154–treated mice (data not shown). Importantly, as determined by immunohistochemical staining, therapy with EC154 significantly reduced tumor cell proliferation (BrdUrd-positive cells) (Figure 4C). Moreover, the ability to inhibit multiple oncogenic signaling pathways in vivo was validated by western blotting of tumor specimens, showing that phosphorylation of AKT and STAT3, as well as expression of IGF1Rβ and EGFR, are diminished upon EC154 treatment (Figure 4E).
Since the microenvironment critically impacts the therapeutic efficacy, we next investigated the impact of EC154 on the growth of orthotopically implanted cancer cells. Indeed, therapy with EC154 significantly reduced pancreatic tumor burden and tumor weight (Figure 5A, and B). Importantly, in vivo expression levels of MST1R were also substantially reduced upon HSP90 blockade with EC154, as compared to controls (Figure 5C). Of note, mouse body weight did not statistically differ among treatment groups (data not shown). From these we conclude experiments that targeting HSP90 with EC154 sufficiently inhibits growth of gastric and pancreatic cancer cells in vivo. In addition, our data provide the first evidence that this inhibition of tumor growth could in part be mediated through the impairement the MST1R system.
Discussion
This study demonstrates that inhibition of HSP90 may directly interfere with MST1R function and MSP1 signaling in both pancreatic and gastric cancer cells. The therapeutic efficacy of targeting HSP90 with the synthetic inhibitor EC154 was validated in subcutaneous and orthotopic tumor models, where EC154 substantially reduced pancreatic and gastric cancer growth. Importantly, inhibition of HSP90 down-regulated MST1R expression in EC154-treated cancer cells both in vitro and in vivo, although MST1R mutations have not been detected in these cancer cells. Moreover, blocking HSP90 elicited functional consequences on MST1R promoter activity. Therefore, our results provide evidence that MST1R represents a new HSP90 client and further supports the rationale of targeting HSP90 in cancer therapy.
The growth-inhibitory effects of HSP90 inhibitors and their interference with oncogenic signaling cascades have been described in various publications (13, 14, 28-30). In this study, we focused on the efficacy of HSP90 blockade, using a novelsynthetic HSP90 inhibitor (EC154) to specifically test whether this inhibitor would disrupt MSP1-activated signaling pathways and affect the oncogenic RTK MST1R. MST1R is a member of the MET proto-oncogene family and activation of MST1R has recently been implicated in the progression and metastasis of various tumor entities, including pancreatic and gastric cancer (6-8, 10, 11, 31-34). Thus MST1R has emerged as an interesting target for cancer therapy and specific inhibitory antibodies are currently under development (33).
In our experiments, blocking HSP90 expectedly led to a substantial inhibition of constitutively activated oncogenic pathways that previously have been investigated in models of gastric and pancreatic cancer (15, 16). Importantly, we now found that HSP90 inhibition also elicits effects on the oncogenic MST1R system. This effect not only involves the interference with MSP1-activated down-stream signaling components, but also MST1R per se in terms of down-regulating MST1R in cancer cells and tumor tissues and reducing its promoter activity (in part via PKC interference). To date, an HSP90-dependency of MST1R has only been shown in one study that used an artificially created constitutively active mutant of MST1R (MST1RM1254T) (17). In this particular study, Germano et al. demonstrated that oncogenic MST1R displays an enhanced physical interaction with HSP90 and increased sensitivity to geldanamycins, resulting in a rapid degradation of MST1RM1254T (17). Therefore, the authors concluded that oncogenic MST1R is an ideal target for the anti-oncogenic activity of HSP90 inhibitors. The fact that wild-type MST1R in our study was susceptible to HSP90 inhibition prompted us to determine the presence of MST1R mutations in our cell lines. Somewhat unexpectedly, we did not detect any mutations, suggesting that wild-type MST1R is a novel HSP90 client protein.
To our knowledge, the effects of HSP90 inhibitors on MSP1/MST1R signaling have not been reported prior to our study. Activation of MST1R through stimulation with its natural ligand MSP1 mediates multiple signaling cascades that are implicated in promoting cell motility, adhesion, proliferation and survival. These MSP1/MST1R signaling components predominantly involve RAS/MAPK and PI3K/AKT (35, 36), as validated in our experiments. Moreover, our results show that blocking HSP90 substantially disrupts MSP1-mediated signaling in pancreatic and gastric cancer cells. This interference occurred in part through inhibition of constitutive and inducible MAPK/ERK1/2 and AKT phosphorylation, as well as through reduction of STAT3 activation. As a functional consequence of such multiple signaling pathway inhibition and MST1R down-regulation, HSP90 blockade by EC154 inhibited MSP1-induced cancer cell migration.
The importance of MST1R in cancer progression has been increasingly investigated over recent years. In this regard, Thomas and co-workers recently reported that MST1R is highly expressed in pancreatic intraepithelial neoplasia, as well as in primary and metastatic pancreatic cancer, suggesting MST1R is involved in pancreatic carcinogenesis (9). These findings are further supported by results from studies by Camp and colleagues, who demonstrated MST1R overexpression in human pancreatic cancer specimens relative to non-malignant ductal tissues (10). Interestingly, a very recent study also provided novel evidence that MST1R may mediate angiogenic activity in pancreatic cancer through modulating vascular endothelial growth factor (VEGF) expression. This represents an important finding since pancreatic cancer tumors excessively produce VEGF and anti-VEGF strategies thus far have failed in the clinic (37). The suitability of MST1R as a therapeutic target has lately been demonstrated in an experimental study that used a novel function-blocking antibody (IMC-41A10) (38). The growth-inhibitory effects of the antibody IMC-41A10 were validated in preclinical tumor models, and a comprehensive analysis was performed on MST1R expression in various human cancer entities. The authors concluded that inhibition of MST1R activity is potentially a useful target for human cancer therapy. However, another recent report has revealed that the growth-inhibitory effects of MST1R inhibitors may be transient (39). In this study, Logan-Collins and co-workers elegantly demonstrated that silencing MST1R reduces tumor growth and renders pancreatic cancer cells susceptible to gemcitabine, however, MST1R down-regulation is lost over time. This transient effect was linked to an increase in c-MET and EGFR expression, suggesting that these two oncogenic receptor systems provide an escape mechanism from MST1R silencing in cancer cells. The EGFR and c-MET findings are particularly relevant to our study since both are HSP90 client proteins. Hence, HSP90 inhibitors may be an effective therapy to achieve a more complete and sustained tumor response, suggesting a new potentially effective antineoplastic concept.
Conclusion
In conclusion, our study demonstrates that MST1R might represent a novel target of HSP90 inhibitors and that MSP1 signaling can be effectively impaired by HSP90 inhibitors. In addition, the novel HSP90 inhibitor EC154 demonstrated strong growth-inhibitory effects in our models. Especially in light of reported escape mechanisms involving c-MET and EGFR using MST1R-directed/silencing therapies (39), our results suggest HSP90 inhibitors may prove to be a more valuable molecular concept in therapy of gastric and pancreatic cancer.
Acknowledgements
The Authors thank Christine Wagner and Eva Scheiffert for excellent technical assistance. These studies were supported in part by the German Cancer Aid (Deutsche Krebshilfe, Max-Eder Programm, Bonn, Germany) (O.S.), and a grant from the University of Regensburg, Medical Faculty (ReForM) (C.M., S.A.L, O.S.).
Footnotes
-
Conflict of Interest
The Authors declare no conflict of interest.
- Received November 30, 2011.
- Revision received December 21, 2011.
- Accepted December 23, 2011.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}