


Abstract
Background: Ribonucleotide reductase composed of the hRRM1 and hRRM2 subunits catalyzes the conversion of ribonucleotides to their corresponding deoxy forms for DNA replication. Anti-hRRM2 siRNA degrades hRRM2's mRNA and suppresses tumorigenesis. A Phase I clinical trial demonstrated its therapy potential. HN-1 represents a tumor-specifically internalizing peptide for targeted-drug delivery into human head and neck squamous cell carcinoma. Materials and Methods: Internalization of peptide was monitored by fluorescence microscopy. The peptide-siRNA conjugate was chemically synthesized. The hRRM2 expression was monitored by western blot analysis. Results: HN-1TYR (HN-1 with two N-terminally added tyrosines) was internalized by human head and neck or breast cancer cells. Anti-hRRM2 siRNAR (resistant to RNase degradation) was conjugated to HN-1TYR without compromising their properties. The treatment with HN-1TYR-anti-hRRM2 siRNAR partly suppressed the endogenously expressed hRRM2 in human breast cancer cells. Conclusion: Our results establish the utility of tumor-specifically internalizing peptides for targeted siRNA delivery into human cancer cells.
Abbreviation: RR, ribonucleotide reductase, TSIP; tumor-specifically internalizing peptide; PKC, protein kinase C; HU, hydroxyurea; HNSCC, head and neck squamous cell carcinoma; PBS, phosphate buffered saline; NHDF, normal human dermal fibroblasts; hRRM2, human ribonucleotide reductase subunit M2; BLAST, basic local alignment search tool; EPR, enhanced permeability retention; siRNA, small-interference RNA.
Ribonucleotide reductase (RR) represents the pharmacological target of the prototypic antimetabolite drug hydroxyurea (HU), which is used to treat human head and neck carcinoma (or sarcoma), chronic myelogenous leukemia and other cancers (1). In mammalian cells, RR represents the sole enzyme catalyzing the reduction of ribonucleotides to their corresponding deoxy forms to provide dNTPs for DNA replication (2). RR is composed of the subunits hRRM1 and hRRM2 (2, 3). HU partly cross-inhibits RR composed of the hRRM1 and p53R2 (a homologue of hRRM2) subunits, which is involved in DNA repair (4). HU inhibits RR by quenching free radicals necessary for the catalysis (2), through generating its own free radicals via oxidative transformation (5). The HU-generated free radicals also react with other molecular entities, resulting in various side-effects (6). HU's short half-life necessitates administering at a higher dose, exacerbating the side-effects.
To avoid HU-induced side-effects, siRNA was used to inhibit RR (7). In a Phase I clinical trial, the siRNA was delivered using nanoparticles that exploit the enhanced permeability retention (EPR) effect to reach the tumors (8). To escalate intravenous drug dose, the diameter of nanoparticles was adjusted to avoid renal clearance (9), though it may trap unused drugs in circulation. The complex was self-assembled from cyclodextrin-containing polymers, polyethylene glycol (for stability), the nucleic acid (siRNA) and human transferrin (to target tumors) (9) though normal cells, like erythrocytes, express the cognate receptor (10). In plasma, beta-cyclodextrins may associate with cholesterol to form crystals in proximal tubule cells and cause nephrotoxicity (11). Upon administration, the siRNA-loaded nanoparticle partly suppressed hRRM2 in melanoma cells of patients (8).
Nevertheless, the above vector carries several limitations. Firstly, the EPR effect occurs inconsistently due to tumor heterogeneity, which may compromise the delivery (12-14). Secondly, its intratumoral distribution was not homogenous (15), as the siRNA-loaded nanoparticle weighs considerably more (~700-10,000 times) than the typical antibodies (~100 kd) that poorly penetrate solid tumors (16). Thirdly, the siRNA-loaded nanoparticle readily disintegrates in the kidney, resulting in rapid depletion (17). Cost-wise, it may be difficult to afford for patients with low-income or in developing countries. These considerations prompted the exploration of alternate delivery vectors. To date, several peptides have been successfully used to deliver siRNAs to tumors in vivo (18). HN-1 (~1 kd) is a tumor-specifically internalizing peptide (TSIP) previously isolated for the targeted-drug delivery into human head and neck squamous cell carcinoma (HNSCC) (19). Using HN-1, various anticancer drugs such as Taxol (20), PKCε-inhibiting peptide (21) and diphtheria toxin (22) or tumor-imaging agents such as radioisotopes (23), have been targeted to human HNSCC in vivo. Here, we describe that the treatment with the HN-1TYR-anti-hRRM2 siRNAR conjugate, partly suppresses the endogenously expressed hRRM2 in human breast cancer cells.

Development of HN-1TYR. Internalization of HN-1TYR by human HNSCC cells. Fluorescence microscopy (phase-contrast optics of the corresponding views are shown in upper panels). Indicated cells were incubated with FITC- HN-1TYR (panels 2 and 4) at 37°C for 48 h, (magnification: ×100).

HN-1TYR targets human breast cancer. Internalization of HN-1TYR by human breast cancer cells. Fluorescence microscopy (phase-contrast optics of the corresponding views are shown in upper panels). Indicated cells were incubated with FITC-HN-1TYR (panels 2, 4 and 6) at 37°C for 48 h (magnification: ×100).
Materials and Methods
Cell lines. All cell lines were acquired from the American Type Culture Collection (Manassas, VA, USA). NHDF, KB, MCF-7, MDA-MB-468 and ZR-75 human cells were maintained in DMEM medium with 10% fetal bovine serum supplemented with antibiotics at 37°C in 10% CO2.
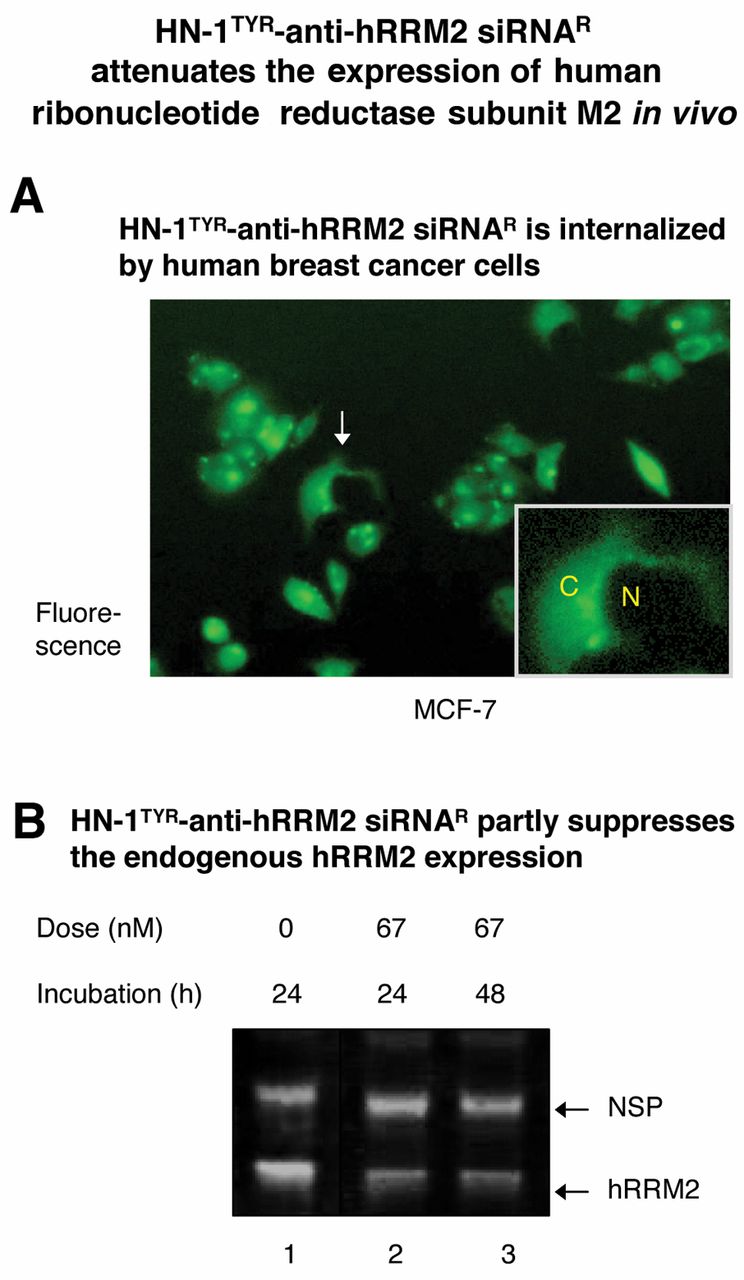
Development of FITC-HN-1TYR-anti-hRRM2 siRNAR. A. Cellular uptake. Human breast cancer MCF-7 cells were incubated with FITC-HN-1TYR-anti-hRRM2 siRNAR (67 nM) at 37°C for 48 h. Fluorescence microscopy was performed as described in Materials and Methods. (Magnification: ×100). Inset: an amplified view of an indicated (arrow) cell [N=nucleus; C=cytoplasm]. B. Suppression of the hRRM2 expression. MCF-7 cells were incubated with FITC-HN-1TYR-anti-hRRM2 siRNAR at 37°C. No transfection agent was used. Western blot analysis was performed as described in Materials and Methods. The band corresponding to hRRM2 or a non-specific protein (NSP) is indicated. Dose: Lane 1, none; lane 2, 67 nM; lane 3, 67 nM. Incubation time: lane 1, 24 h; lane 2, 24 h; lane 3, 48 h.
Fluorescence microscopy. Cells were placed in 8-chamber slides and incubated in the appropriate medium, as described (19). After treating with the indicated peptide, cells were fixed, mounted, and viewed using an Olympus fluorescence microscope at the Microscope Core Facility of City of Hope National Medical Center (Duarte, CA, USA).
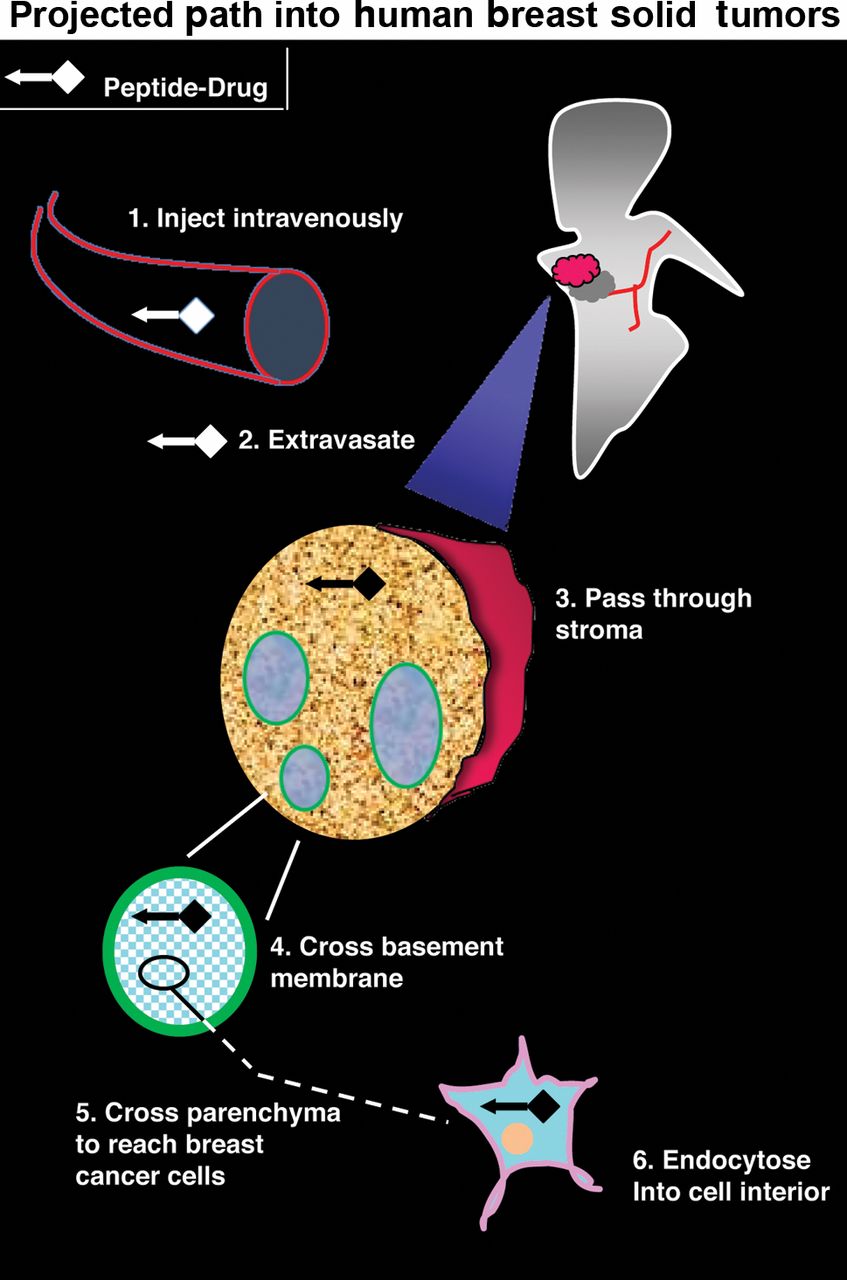
Map of the peptide-drug trajectory. A schematic representation shows the projected path of a peptide-drug conjugate into human breast solid tumors.
Western blot analysis. Western blot analysis was performed using anti-human hRRM2 subunit antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) as the primary antibody, followed by alkaline-conjugated anti-mouse IgG antibody as the secondary antibody. The resulting blot was visualized via the enhanced chemilumescence method (24).
Peptide or siRNA synthesis. Peptide, siRNA, or peptide-siRNA was synthesized and purified to >95% at the Peptide Synthesis Core Facility of City of Hope National Medical Center (Duarte, CA, USA). The predicted molecular mass and purity was confirmed by mass spectrometry. For FITC-HN1 (TSPLNIHNGQKL) or FITC-HN-1TYR (YYTSPLNIHNGQKL), a fluorescent label was added at the NH2-terminus and the COOH-terminus was capped with an amide group. FITC-HN-1TYR-anti-hRRM2 siRNAR was prepared as described in text (see below).
Results and Discussion
Development of the targeted-delivery vector HN-1TYR. HN-1, a synthetic 12-mer TSLP, was isolated by screening a random peptide-displaying M13 bacteriophage-based library (19). It meets several criteria for targeted drug delivery into solid tumors. Firstly, HN-1 can translocate drugs across the cell membrane into the cytosol. This was originally inferred from the HN-1 internalization study performed using a fluorescent molecule as the cargo (19). More recently, HN-1 was shown to translocate a human protein kinase C-ε (PKC-ε) inhibiting peptide or diphtheria toxin used as therapeutic into human cancer cells (21, 22). Secondly, the uptake of HN-1 occurs in a tumor-specific manner. HN-1 is internalized by human HNSCC cells (i.e. MDA177Tu, MDA138Tu, MDA59Tu, MDA167Tu, MDA686Tu, MDA1986Tu, UMSCC1, UMSCC36) yet poorly by their normal counterparts (i.e. HOK16B, NOE oral epithelial cells) (19, 21). Similarly, HN-1 was internalized by human breast cancer cells (ex. MDA-MB231, SKBR3) but less efficiently by their normal counterpart (ex. MCF10A non-tumorigenic mammary epithelial cells) (21). Thirdly, HN-1 is capable of penetrating solid tumor, which is critical as >90% of human cancers represent solid tumors. This was indicated by the presence of intravenously injected HN-1 at the interior of solid tumors formed from MDA177Tu or MDA167Tu cells in a mouse xenograft model (19). An independent report confirmed that the intravenously injected HN1 localizes at the interior of solid tumors formed from UMSCC1 cells in vivo (21).
HN-1TYR, which consists of the parental HN-1 backbone with two extra tyrosine residues, was synthesized. Tyrosines were placed N-terminally as the presence of additional residues at this terminus did not interfere with the HN-1 internalization potential (19). HN-1TYR efficiently solublized in phosphate-buffered saline (PBS) solution. The addition of two tyrosines did not generate novel proteolytic sites. Next, human head and neck cancer KB cells were incubated with FITC-HN-1TYR at 37°C, for 48 h, as previously described (19). FITC-HN-1TYR, which represents fluorescein-conjugated HN-1TYR, was synthesized chemically (see Materials and Methods). It led to fluorescing KB cells. Fluorescence was detected throughout the cell and was not limited to the cell surface (Figure 1; panel 2; also see Figure 2). The enlarged fluorescent micrograph of a single cell (Figure 3A, inset) shows that the internalized peptide primarily resides at the cytoplasm consistent with the properties of the parental peptide HN-1 (19). In the original report describing the HN-1 isolation (19), its internalization was extensively documented using various experimental methods so it is not repeated here. Untreated KB cells exhibited little autofluorescence (Figure 1; panel 1), suggesting that the observed fluorescence (Figure 1; panel 2) was specifically due to the internalized FITC-HN-1TYR. In contrast, normal human dermal fibroblasts (NHDF) that were similarly incubated with FITC-HN-1TYR exhibited little fluorescence (Figure 1, panel 4). Thus, HN-1TYR retains the HNSCC-internalization potential.
HN-1TYR is internalized by human breast cancer cells. To identify additional cancer targets of HN-1 with the paucity of information regarding the cognate receptor, we relied on the common developmental lineage. Previously, a similar approach (i.e. ‘developmental histogenesis’) was used to identify cancer types with similar chemosensitivity (25). During embryonic development, surface ectodermal epithelium gives rise to three distinct structures—i.e. cranial structures, epidermal structures, ectodermal placodes (26). Cranial structures include multiple cell types within the oral cavity such as the secretory, duct-lining cells of the salivary, palatine of oral glands and the epithelial lining cells of the gums, palate, lips and paranasal sinuses. Epidermal structures include the duct-lining and secretory cells of mammary glands (27). In humans, breast cancer occurs most frequently in the duct (i.e. ductal carcinoma) or lobules (i.e. lobular carcinoma). This fact, along with the recent finding by Bao et al. that HN-1 is internalized by human breast cancer cells (21), prompted us to investigate whether HN-1TYR could be used to deliver drugs to breast cancer cells.
To examine this point, human breast cancer cells with varying ‘triple status’ were investigated. Triple status refers to the expression level of Her2 receptor, estrogen receptor (ER), and progesterone receptor (PR). Here, we used human breast cancer cells that are distinct from those used in the report by Bao et al. (21). ZR-75-1 cells, which overexpress Her2, are positive for the ER (16 fmol/mg protein) or PR (102 fmol/mg) expression (28, 29). MCF-7 cells, which express little Her2, are positive for the ER (43 fmol/mg protein) or PR (115 fmol/mg protein) expression (30-32). MDA-MB-468 cells, which express little Her2, contain little ER (4 fmol/mg protein) or PR (9 fmol/mg protein (28, 33). Incubation of ZR-75-1 (Figure 2, panel 4), MCF-7 (Figure 2, panel 6) or MDA-MB-468 (Figure 2, panel 2) with FITC-HN-1TYR at 37°C for 48 h led to fluorescing cells in all three cases. In contrast, untreated ZR-75-1 (Figure 2, panel 3), MCF-7 (Figure 2, panel 5) or MDA-MB-468 (Figure 2, panel 1) cells did not fluoresce, indicative of the lack of autofluorescence. Hence, like HN-1, HN-1TYR targets human breast cancer cells and the targeting may be independent of their triple status.
Development of HN-1TYR-anti-hRRM2 siRNAR for anticancer therapy. To avoid side-effects resulting from the HU chemotherapy, an anti-hRRM2 siRNA that suppresses RR by degrading the mRNA encoding its hRRM2 subunit was previously developed (7). To identify target site, the hRRM2 mRNA sequence was probed for the preferred 27-nucleotide sites using an algorithm designed for predicting end-energy differential and mRNA secondary structure (34). The resulting candidate were distinguished on the basis of their GC content and homology with non-hRRM2 genes using basic local alignment search tool (BLAST) search, which yielded 3 siRNA candidates. Subsequently, numerous duplexes in their immediate vicinities were analyzed, resulting in the identification of a single siRNA with a potent hRRM2 inhibiting property (7). Upon transfection (using lipofectamine), the anti-hRRM2 siRNA suppressed (40-60%) the hRRM2 protein level for as long as 5 days. Transfection of human colorectal adenocarcinoma HT-29 cells with the anti-hRRM2 siRNA led to a near complete cessation of their growth (7). The siRNA does not appear to elicit interferon response in vivo (8, 35).
To translocate across the cell membrane for cellular uptake, the above siRNA was conjugated to HN-1TYR. To avoid degradation by RNases in vivo, anti-hRRM2 siRNA was synthesized with fluorine, incorporated at its 2’-OH position to yield anti-hRRM2 siRNAR. FITC-HN-1TYR was conjugated to 5’-end of anti-hRRM2 siRNAR (antisense strand only) using hexynyl phosphoramidite linker to generate FITC-HN-1TYR-anti-hRRM2 siRNAR. Equimolar concentration of the above sense and antisense strands were hybridized to generate duplex siRNA (molecular mass of the conjugate: ~19.7 kd). The internalization potential of the conjugate was examined. MCF-7 breast cancer cells were incubated with FITC-HN-1TYR-anti-hRRM2 siRNAR at 37°C for 48 h, as described above. It led to fluorescing cells (Figure 3A). The inset shown in Figure 3A, representing the amplified view of a single treated cell, shows clearly that the conjugate localizes at the cytoplasm upon entry. Untreated MCF-7 cells did not fluoresce (panel 5 in Figure 2). Thus, HN-1TYR is capable of translocating anti-hRRM2 siRNAR into MCF-7 cells in vivo. Next, its effect on the hRRM2 expression was examined. MCF-7 cells were incubated with FITC-HN-1TYR-anti-hRRM2 siRNAR for the indicated time period, lysed, and the resulting cell lysate was analyzed by western blot analysis using an anti-human hRRM2 antibody as the primary antibody. It led to a partial reduction (ca. ~50%) in the level of the endogenously expressed hRRM2 (Figure 3B, lanes 2 or 3). The suppression was specific for hRRM2 as the expression level of an irrelevant protein (designated as “NSP” in Figure 3B, lanes 2 or 3) was not affected. Thus, the FITC-HN-1TYR-anti-hRRM2 siRNAR conjugate retains the hRRM2-suppressing property.
A key determinant for the successful siRNA therapy is delivery (36). To avoid side-effects, delivery vectors with tumor-homing potential are increasingly sought. For targeted-drug delivery into human solid tumors, multiple requirements must be met (see above). Minute, non-bioactive, tumor-specifically internalizing peptides such as HN-1 may provide a solution. In this report, we described the development of a conjugate composed of the HN-1TYR peptide and anti-hRRM2 siRNAR. Our results confirm earlier findings by others that peptides can mediate the transfer of siRNAs into human cells (18). As the uptake seems to occur independently of the ‘triple status’, it may be applicable for a wide spectrum of human breast cancers. The results also suggest that RR remains a tenable target for therapy. Novel chemotherapeutics inhibiting RR are currently being developed. Potentially, siRNAs may represent the next-generation drugs to target RR. Its specificity may be exploited to lessen side-effects while preserving their therapeutic potential. Further works are envisioned. Firstly, despite the recent report (37), the precise identity of the HN-1 receptor remains to be defined. Secondly, a HN-1 derived vector with the endosome-escaping potential may be developed (22). Thirdly, the tumor-suppressing potential of the conjugate will be examined using an animal model. Hopefully, these advances may lead to a safer, yet effective, form of anticancer therapy.
Acknowledgements
We thank Xiyong Liu and Shuya Hu for technical assistance, and C. Bassford, D. Drake and M. Kong for administrative assistance.
- Received September 12, 2012.
- Accepted September 24, 2012.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.