


Abstract
Background: The tumour suppressor gene ‘mutated in colorectal cancer' (MCC) is silenced through promoter methylation in colorectal cancer and has been implicated as a regulator of the nuclear factor kappa B (NFκB) pathway. Therefore, we aimed to determine whether MCC modulates NFκB activation in colorectal cancer. Materials and Methods: NFκB activation was assessed using luciferase reporter assays in colorectal cancer cells in vitro. MCC methylation was analysed in primary tumour specimens from patients with inflammatory bowel disease. Results: Re-expression of MCC reduced NFκB-dependent transcription in tumour necrosis factor alpha (TNFα)- or lipopolysaccharide (LPS)-stimulated cells. Conversely, knockdown of MCC resulted in accumulation of the inhibitor of kappa B alpha (IκBα) protein, encoded by NFKBIA, a first response gene specifically and rapidly regulated by NFκB pathway activation. The MCC gene is methylated in up to 6/16 of inflammatory bowel disease-associated tissue specimens, and myosin-10 and valosin-containing protein were identified as MCC-interacting proteins. Conclusion: These findings suggest that MCC modulates NFκB pathway signalling indirectly in colorectal cancer cells.
The MCC gene was discovered during the search for the familial adenomatous polyposis locus because of its close linkage to the APC gene (1). However, as MCC is only rarely mutated in colorectal cancer (CRC) (2), its significance in carcinogenesis was initially poorly understood. It was recently reported that the MCC gene promoter is methylated in up to 50% of CRCs and 80% serrated polyps, suggesting that its silencing is important in early colon carcinogenesis via the serrated neoplasia pathway (3, 4). Furthermore, an unbiased genetic screening of a mouse model of CRC implicated MCC mutation as a key event in colorectal carcinogenesis (5), providing evidence that it is a tumour suppressor gene. These findings have renewed interest in the cellular function of MCC.
MCC has been implicated as a negative regulator of the cell cycle and cell proliferation (6). We have confirmed the role of MCC in cell cycle regulation in CRC cells and shown that MCC protein phosphorylation is required in response to DNA damage caused by single-strand DNA breaks (7). MCC has also been implicated as a modulator of the nuclear factor kappa B (NFκB) signalling pathway through its interaction with the inhibitor of kappa B (IκB), the cytoplasmic inhibitor of NFκB signalling (8). However, the functional significance of this interaction in CRC was not addressed in this large-scale mapping study of NFκB–protein interactions. Thus, the potential interplay between MCC and NFκB signalling has yet to be determined, in particular in CRC, where MCC is frequently silenced by promoter methylation.
The NFκB signalling pathway is important in the normal development of the colorectum and maintenance of colonocyte viability, but is aberrantly activated in CRC (9-11). Patients with inflammatory conditions, such as ulcerative colitis, exhibit increased NFκB activity in colonocytes and have a greater risk of developing CRC (12). NFκB activation in both epithelial and myeloid cells contributes to CRC and this has been described as an important link between inflammation and cancer (13, 14).
Numerous NFκB activators have been described, including a diverse array of ligand–receptor interactions. Rapid canonical NFκB signalling by inflammatory factors is mediated mainly by members of the interleukin and tumour necrosis factor receptor (TNFR) protein superfamilies, the best characterized being TNFR1 (15, 16). Transduction of the initiating signal, such as TNFα or lipopolysaccharide (LPS), is mediated by intracellular proteins, which induce a signalling cascade culminating in activation of the IκB kinases (IKK), IKKα or IKKβ. These kinases phosphorylate NFκB cytoplasmic inhibitors IκBα and IκBβ, leading to the nuclear translocation and activation of NFκB transcription factors RelA or RelB.
Therefore, due to the strong association between increased NFκB activity and the predisposition to CRC, the high frequency of MCC methylation in early colorectal polyps, and the reported interaction between MCC and IκBβ, the effect of MCC expression on NFκB signalling was further examined.
Materials and Methods
Patient cohort and MCC methylation analysis. Colorectal carcinoma specimens from individuals with inflammatory bowel disease were drawn from patients who had undergone complete surgical resection of a primary CRC between 1993-2010. The 16 individuals (9 male and 7 female) were aged 31 to 82 years. Genomic DNA was extracted from cancer and the matching non-cancer colonic mucosa. Quantitative real-time PCR for MCC gene promoter methylation was carried out using an ABI7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA) and myogenic differentiation 1 (MYOD) as the reference gene as previously described (3, 17).
Construction of plasmids. Full-length MCC cDNA (2490 bp) was amplified by PCR from cDNA clone MGC:12731 (Invitrogen, Carlsbad, CA, USA) using extended attB primers (forward 5’-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGAATTCCG GAGTTGC-3’; reverse 5’-GGGGACCACTTTGTACAAGAAAGCT GGGTATTAAAGCGAAGTTTCATT-3’) and cloned into the Gateway entry vector pDONR221 (Invitrogen). Wild-type MCC was subsequently recombined into pcDNA3.1/nV5-DEST (Invitrogen) using the Gateway® LR reaction as per the manufacturer's instructions.
Protein analysis. Cell lysates were generated using RIPA buffer supplemented with protease inhibitors (3), and proteins (10-30 mg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 8% acrylamide gels. The following antibodies were used for Western blot analysis: from Cell Signaling Technology (Danvers, MA, USA) AKT (#9272), phospho-AKT (#9271), ERK1/2 (#4696), phospho-ERK1/2 (E10, #9106), IκBα (#9242), phospho-IκBα (#14D4, #2859), IκBβ (#9248), phospho-IκBβ (#4821), IKKβ (#2684), phospho-IKKα/β (16A6, #2697), RelA (#3034), phospho-RelA (93H1, #3033), as well as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (4300; Ambion, Austin, TX, USA), myosin-10 (M7937; Sigma, St Louis, MO, USA), nV5 (46-0705; Invitrogen), VCP (sc-20799; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and MCC (610740; BD Transduction Laboratories™, Franklin Lakes, NJ, USA).
Mass spectrometry (NanoLC ESI MS MS) was performed using Agilent 1100 Series (Agilent Technologies, Boeblingen, Germany) and QStar XL (Applied Biosystems, Framingham, MA, USA) instruments. Peptides were assigned by searching the SWISS-PROT human protein database using Mascot (Matrix Sciences, London, UK). MCC-binding partners were confirmed by standard immunoprecipitation (IP) method using the Tris, Sodium Chloride, EDTA (TNE) buffer system, followed by Western blot.
Luciferase activity assays. KM12SM and HCT-15 (CCL-225™; American Type Culture Collection, ATCC, Manassas, VA, USA) cells were transfected in triplicate with a NFκB-luciferase reporter (0.66 μg) (18), a lacZ/beta-galactosidase expression plasmid to control for transfection efficiency (0.33 μg; pEF/GW-51/lacZ; Invitrogen) and either 1.5 μg of empty pcDNA3.1/nV5-DEST or the MCC-containing equivalent vector. Transfection medium was replaced with serum starvation (SS) medium (0.2% fetal calf serum, FCS) 24 h post-transfection, and the cells serum-starved for a further 16 h. To determine changes in NFκB activation, cells were stimulated with either TNFα (10 ng/ml, CytoLab, Rehovot, Israel) or LPS (10 μg/ml, Invivogen, San Diego, CA, USA) for 6 h. Lysates were harvested and NFκB-luciferase and β-galactosidase activity was determined using the Galacto-Star™ System (Tropix Inc, Bedford, MA, USA). Relative luciferase activity for a sample was determined by dividing the average NFκB-luciferase activity, by the relative amount of β-galactosidase activity.
Treatment with siRNAs. SW480 (CCL-228™; ATCC) and HEK293 (CRL-1573™; ATCC) cells were transfected using Lipofectamine2000 with 10 nM ON-TARGETplus siRNA from Dharmacon (Lafayette, CO, USA): MCC siRNA #9 (J-010523-09), MCC siRNA #10 (J-010523-10), non-targeting #2 control siRNA (D-001810-02-20) and RNA-induced silencing complex (RISC)-free control siRNA (D-001220-01-20), as per the manufacturer's instructions. Opti-mem (31985, GIBCO®; Invitrogen) was used in place of serum-free media. siRNA concentration of 10 nM resulted in a reduction of MCC expression between 80-90% compared to controls.
Caspase-8 activation and global apoptosis assays. HCT-15 cells were transfected with 1.5 μg empty vector, or wild-type MCC-expressing construct. Twenty-four hours post-transfection, cells were cultured in SS medium for 16 h, followed by stimulation with TNFα (10 ng/ml in 5% FCS medium) for 24 h. Caspase-8 activation was assayed from floating and adherent cells (Apoptosis Detection Kit – Caspase-8; Kamiya Biomedical Company, Seattle, WA, USA) and the covalently-bound fluorescent caspase-8 inhibitor was detected using the FL-1 channel by flow cytometry. Caspase-8 activation was determined by comparisons of the number of fluorescent cells before and after TNFα treatment. To determine global apoptosis, floating and adherent cells were collected from empty vector or MCC-expressing HCT-15 cells were subjected to the same TNFα treatment used for luciferase assays, and the DNA content determined by staining with propidium iodide and analysis by flow cytometry.

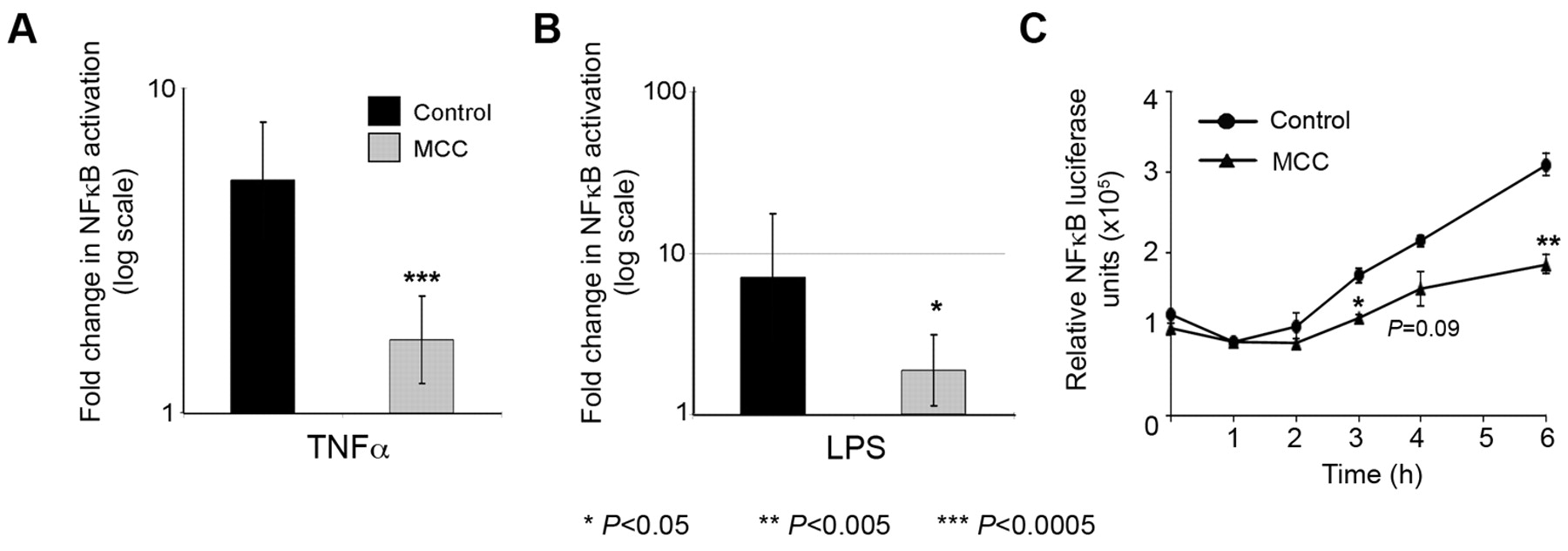
MCC modulates NFκB-mediated transcription. KM12SM cells (A, B) and HCT-15 cells (C) were transfected in triplicate with the NFκB reporter, a lacZ transfection control plasmid, and either MCC.pcDNA3.1/nV5-DEST or empty vector control for 24 h. Cells were cultured in serum starvation (SS) medium (0.2% FCS), or SS medium supplemented with TNFα (10 ng/ml) or LPS (10 μg/ml) for 6 h prior to harvesting (A, B) or harvested hourly at 1-6 h (C).
Statistical analysis. Paired t-tests were used to determine the level of significance between experimental groups (Excel). P-values for differences between fold-change across three separate luciferase experiments were generated by performing a two-sided paired t-test, assuming equal variance as indicated by the data after log transformation.
Results
MCC gene is methylated in a subset of inflammatory bowel disease-associated tissue specimens. In order to investigate a possible role of the MCC defect in inflammation-associated CRC, we first analyzed a series of primary tumours resected from inflammatory bowel disease patients. MCC methylation was detected in 6/16 of the tumours and in 3/16 of the matching non-cancer tissue specimens. In one patient, both specimens showed MCC methylation, but in the others, methylation was only found in either the cancer or the matching non-cancer tissue from the inflamed colon. In contrast, MCC methylation was not observed in the normal colon of healthy controls (3).
MCC modulates NFκB transcriptional activity in colorectal cancer cells. The effect of MCC re-expression on NFκB-dependent transcription was investigated in serum starved KM12SM cells in which MCC expression is abolished due to methylation of the promoter (3). Pathway-dependent transcription was measured by luciferase activity after co-transfection of MCC or control vectors and a NFκB luciferase reporter in conditions where the NFκB pathway is activated. Transient MCC expression consistently reduced the magnitude of NFκB activation in response to both TNFα and LPS in three separate experiments (Figure 1A and B; p=0.0003 and p=0.0127, respectively). Similar results were obtained for a second MCC-null CRC cell line, HCT-15. A time-course experiment showed that the inhibitory effect of MCC re-expression was detectable after 3 h of TNFα treatment (Figure 1C).

MCC overexpression reduces levels of key NFκB signalling proteins. KM12SM cells were transfected with MCC or an empty vector control for 24 h. Cells were cultured for a further 16 h in full medium or SS medium, followed by a 6 h incubation in full, SS, or SS media supplemented with TNFα (10 ng/ml) or LPS (10 μg/ml). Proteins (20 μg) were separated by SDS-PAGE and subjected to western blot. Primary antibodies were incubated overnight, and protein levels visualized by chemiluminescence. GAPDH was used as the protein loading control.
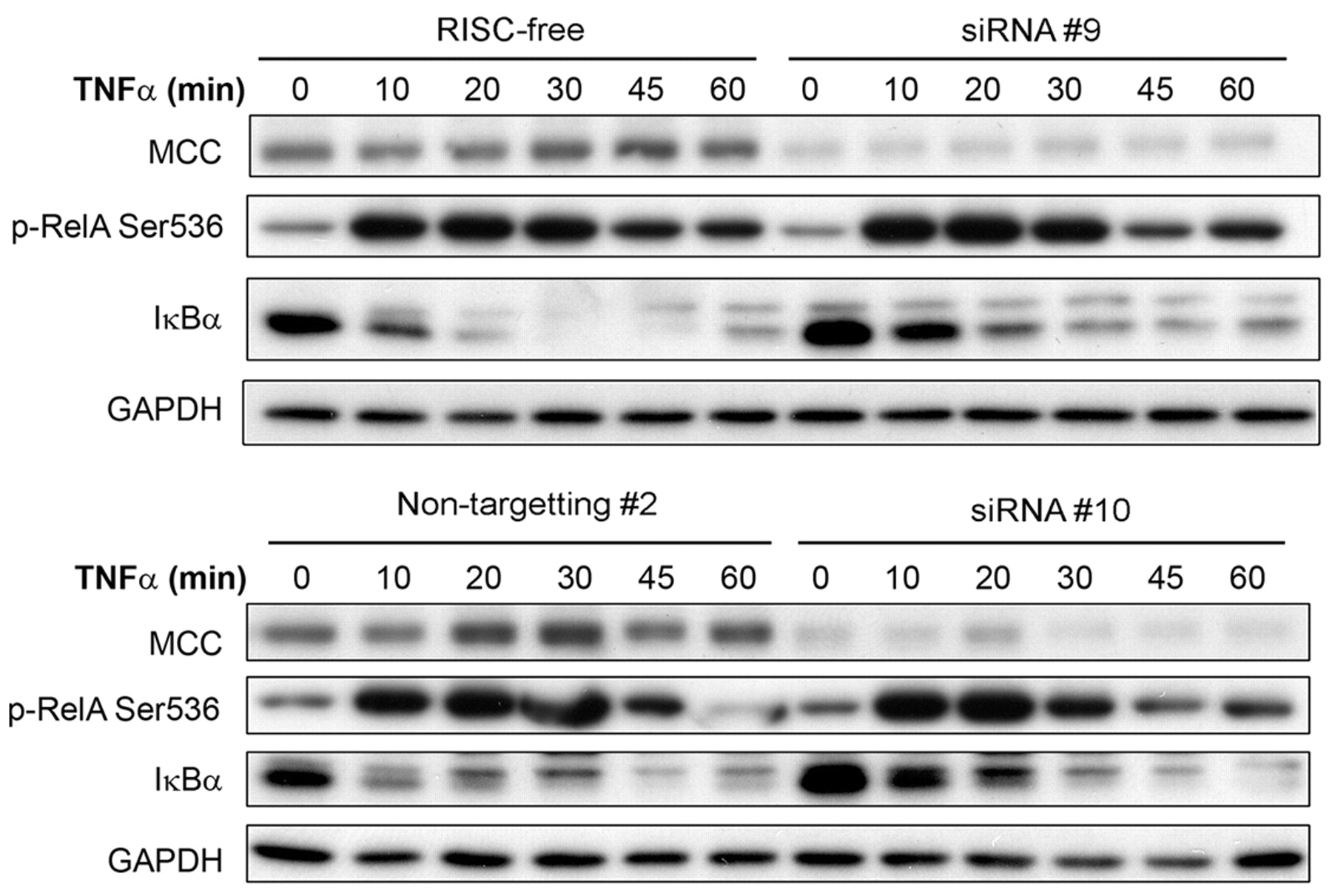
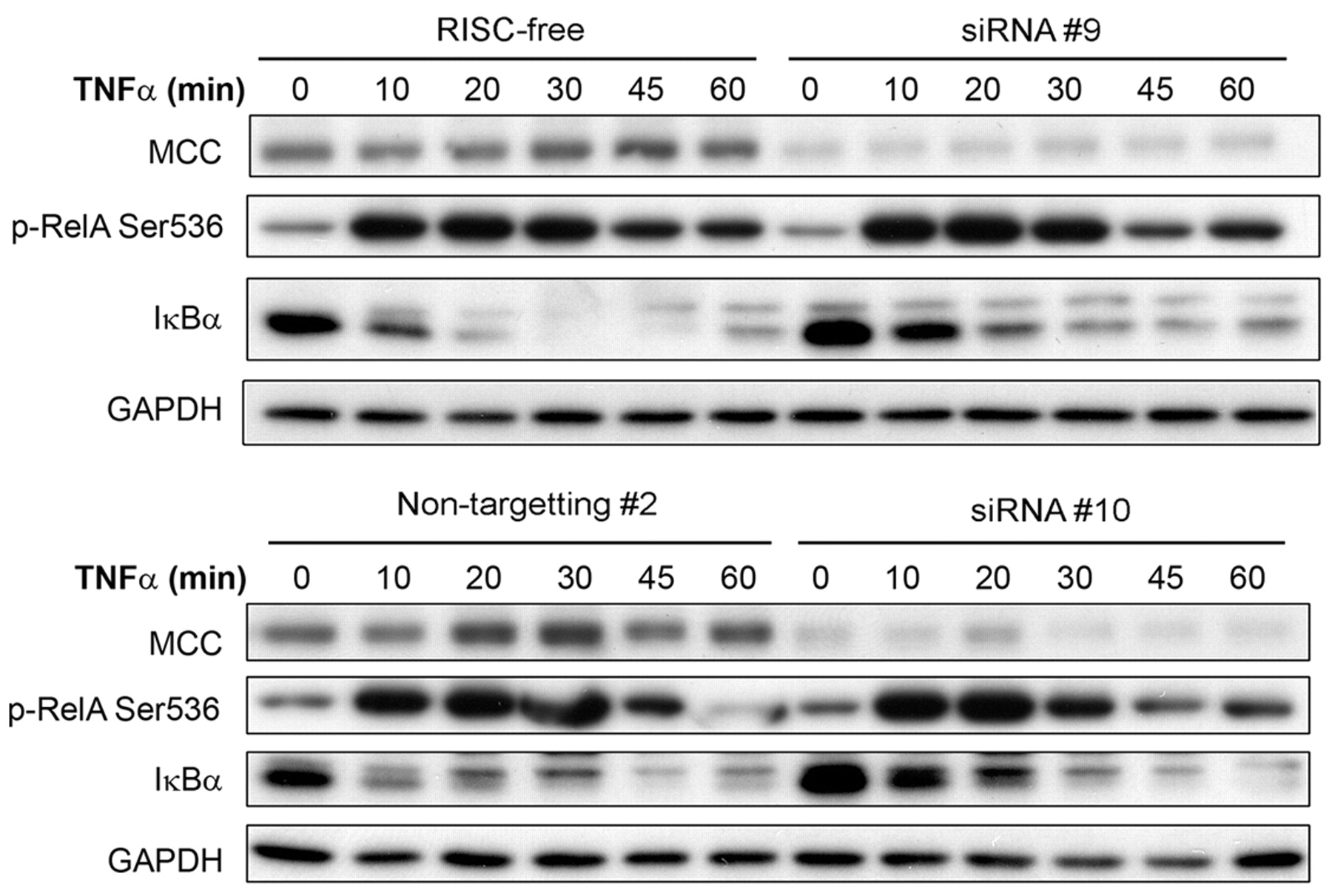
MCC knockdown increases IκBα levels on TNFα stimulation. HEK293 cells were transfected with 10 nM MCC-specific siRNAs (siRNA #9 and siRNA #10), or control siRNAs (RISC-free and Non-targeting #2) for 24 h, followed by culture in SS medium for 16 h. Cells were stimulated with TNFα (10 ng/ml) and protein lysates collected at the time points indicated. Proteins were separated by SDS-PAGE and subjected to Western blot. GAPDH was used as the protein loading control.
MCC re-expression decreases the levels of NFκB pathway proteins. Because MCC re-expression reduced NFκB transcriptional activity in response to both TNFα and LPS, the protein levels of major NFκB pathway signalling proteins were examined. MCC re-expression in KM12SM cells caused a 40-50% decrease in the total and phosphorylated protein levels of the NFκB pathway components RelA, IκBα and IκBβ after 6 h of TNFα, and after LPS treatment (Figure 2). In serum-starved cells, there was a similar small decrease of phosphorylated, as well as total RelA, IκBα and IκBβ with MCC re-expression. Down-regulation of total protein levels was specific for NFκB signalling proteins, as AKT (PI3K signalling), and both ERK1 and ERK2 (MAPK signalling pathway) protein levels remained relatively unchanged (Figure 2). Conversely, knockdown of MCC levels in HEK293 cells resulted in an accumulation of IκBα protein (Figure 3), encoded by NFKBIA, a first response gene specifically and rapidly transcribed by NFκB (19). When stimulated with TNFα for 60 min, coincident RelA phosphorylation and degradation of accumulated IκBα was observed in HEK293 cells with normal MCC expression (Figure 3). With MCC knockdown, RelA phosphorylation was unchanged in response to TNFα stimulation, but there was a delay in IκBα protein degradation, indicating increased IκBα transcription and NFκB activity.
MCC re-expression does not increase global apoptosis. Because a reduction in NFκB signalling can result in increased apoptosis, we tested whether MCC-induced NFκB inhibition could affect the number of cells undergoing TNFR1-mediated apoptosis. The number of cells with activated caspase-8, an essential initiator of TNFR1-associated apoptosis, increased in the presence of TNFα, indicating a functional TNFR1-mediated apoptosis pathway in this cell line. However, MCC re-expression did not further increase TNFα-mediated apoptosis compared to vector-transfected control cells (24.2% vs. 30.9% of TNFα treated cells; Figure 4A and B), thus the presence of MCC did not promote a pro-apoptotic response during chronic TNFα stimulation. Furthermore, an increase in the number of cells exhibiting DNA fragmentation, a marker of global apoptosis, was not observed in MCC-overexpressing cells compared with controls in both unstimulated (12.1% vs. 11.8%; Figure 4C and D) and TNFα treated cells (15.4% vs. 15.8%; Figure 4E and F).
MCC interacts with myosin-10 and valosin-containing protein. To gain further insight into how MCC may exert its suppressive effect on NFκB signalling, proteins that co-immunoprecipitated with endogenous MCC from HEK293 were identified by mass spectrometry (Table I). We did not observe the previously reported MCC interaction with IκBβ (8) but identified myosin-10 (MYH10), which was previously detected in a large-scale study of protein–protein interactions as an MCC-binding partner (20). In additional mass spectrometry experiments, FLAG-tagged MCC was immunoprecipitated with a FLAG antibody from SW480 colorectal cancer cells (7). This detected another MCC-interacting protein that was found in the large-scale study, valosin-containing protein (VCP). Subsequently, we confirmed the interaction of MCC with MYH10 and VCP in KM12SM colorectal cancer cells overexpressing MCC using co-immunoprecipitation (Figure 5) but again, an interaction with IκBβ was not detected.

Re-expression of MCC does not promote apoptosis. Cells with activated caspase-8 were detected and counted by flow cytometry using the FL1-H channel. The empty curves represent histogram counts from unstimulated HCT-15 cells, shaded curves for TNFα-treated cells, and percentages shown are the proportion of the population contained within the active caspase-8 gate of control (A), and MCC-expressing cells (B). Global apoptosis was determined flow cytometry by identification of the sub-G1 DNA content population (%) in MCC (D, F) and control (C, E) transfected cells. Transfected cells were subjected to culture in SS medium (C, D) and SS medium supplemented with TNFα (10 ng/ml; E, F) for 6 h.
Discussion
MCC is emerging as a multifunctional protein that affects several cellular processes and pathways, such as NFκB (8), Wnt signalling (4) and the cell cycle (6, 7). Re-expression of MCC inhibits cell proliferation in several cancer cell lines (4, 6, 7). However, it is poorly understood how the loss of MCC promotes carcinogenesis in the colon.

Confirmation of the MCC protein interaction with the MYH10 and VCP proteins. KM12SM cells were transfected with MCC or control vectors for 8 h, and cultured in media containing 10% serum for a further 40 h. Lysates were generated using TNE/0.5%Triton-×100, and immunoprecipitates were generated from total protein (10 mg) by incubation for 2 h with 2 μg antibody and isolation using protein-G-sepharose 2B beads. Entire samples were loaded, and proteins separated by SDS-PAGE and subjected to Western blot analysis.
The highest ranking proteins that immunoprecipitated with the MCC antibody in HEK293 cells, detected with mass spectrometry. Candidate MCC-interacting proteins, which were not detected in negative controls, are shown in bold.
MCC gene is silenced through promoter methylation in CRC and in precancerous polyps (3, 4). The reported frequency of MCC methylation in sporadic CRC is 40-50%. To date, MCC methylation has not been described in any other type of cancer. Here we show for the first time that the MCC gene is methylated in 6/16 of inflammatory bowel disease-associated cancers and 3/16 of the adjacent control tissues. It is possible that the flat tissue in the inflamed colon of these patients contains dysplastic regions, which may explain the presence of MCC methylation in the matching non-cancer specimens. We have previously observed MCC methylation in premalignant sporadic polyps, but not in the normal colon.
NFκB activation is central to the neoplastic transformation of epithelial cells in the context of chronic colon inflammation (13, 14). Therefore, loss of factors that normally inhibit NFκB signalling may contribute to the disease progression or carcinogenesis in chronic inflammatory bowel disease. This study describes a new role for MCC as a transcriptional modulator of the NFκB pathway in CRC cells, which was previously suggested in HEK293 human embryonic kidney cells (8). While re-expression of MCC reduced activation of NFκB signalling in response to TNFα and LPS, it did not result in increased caspase activity or apoptosis in CRC cells despite specific reduction in key NFκB pathway signalling protein levels. An increase in IκBα was observed on MCC knockdown with siRNA, which suggests that MCC depletion increased NFκB pathway activity, as NFKBIA is a gene rapidly induced by NFκB activity due to three NFκβ response elements present in the promoter region (19). NFKBIA plays a self-limiting and critical role in the negative feedback NFκB pathway signalling.
MCC has previously been found in complex with IκBβ, which together with IκBα, binds to RelA preventing its translocation to the nucleus and transcription of NFκB target genes (8). While MCC binding to IκBβ would potentially explain how MCC modulates the NFκB pathway (8), we could not confirm this interaction. However, we identified other binding partners for MCC in IP mass spectrometry experiments, including other possible regulators of NFκB signalling. There were similarities between the MCC-interacting proteins identified in this study and a previous study by Ewing et al. (20), which used MCC as a ‘bait’ protein. Two proteins, MYH10 and VCP, were identified in both studies suggesting a robust interaction. These interactions were confirmed here by co-IP in CRC cells. Ewing et al. placed MYH10 and VCP in the top ten most robust MCC interactions as determined by the interaction confidence scores (20). Both VCP and MYH10 have been implicated in NFκB signalling through multiple protein–protein interactions. VCP interacts with IκBα, IκBβ and IκBε, MYH10 with RelA, IKKβ and NEMO and both proteins interact with IKKε (8, 20).
Apart from large-scale protein–protein interaction studies, there is some evidence for functional interactions of MYH10 and VCP with NFκB signalling. MYH10 is a structural part of the active cytoskeleton, which is rearranged during TNFα-induced RelA nuclear translocation (21). VCP can affect NFκB activation directly through binding to cytoplasmic IκBα causing its degradation, which is required for the nuclear translocation of NFκB transcription factors (22). It has been suggested that this provides a cellular mechanism whereby aberrant VCP activity promotes tumour metastasis in an experimental osteosarcoma model (23). In colorectal carcinoma, high expression of VCP is associated with poor prognosis and disease recurrence (24).
MCC is emerging as a multifunctional protein that can affect the transcription of multiple pathways including Wnt and NFκB. The functional diversity of MCC binding partners suggests that MCC may have diverse roles in the colon. Mutated MCC has been shown to be a driver of carcinogenesis in a mouse model of CRC (5), indicating that an MCC defect is functionally important in early lesions (3). MCC silencing may also be important in a subset of inflammation-associated CRC.
Acknowledgements
The Authors thank the Cancer Institute NSW, Cancer Council NSW and the Australian Cancer Research Foundation for funding, Nicola Currey and Allison Arndt for technical assistance. MKC and EAM are Cancer Institute NSW Career Development Fellows and NDS is a recipient of the University of NSW Postgraduate Award.
- Received October 5, 2011.
- Revision received December 1, 2011.
- Accepted December 2, 2011.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}