


Abstract
Current attempts to disrupt the complex process of tumor blood vessel formation are predominantly focused on targeting the vascular endothelial growth factor (VEGF)–VEGFR signaling pathway. Although clinically proven to inhibit VEGF and its receptors, these pharmacologic agents are selective, but not specific. Consequently, many of the approved inhibitors also impair other molecular targets leading to increased toxicity. Current efforts to unravel the complexity of tumor angiogenesis have identified several new candidates for antivascular therapy. In this review article, we identify well-established and novel angiogenic molecules and discuss benefits of the therapeutic approaches based on targeting of such factors.
Destruction of the tumor vasculature has long been accepted as a potential strategy for controlling tumor growth. Due to its critical role in tumor oxygenation and the delivery of growth factors, hormones and nutrients, tumor blood vessel formation (i.e. tumor angiogenesis) has been identified as a fundamental process for the maintenance and progression of many solid tumors. The development of tumor vasculature is modulated by a highly complex regulatory network of pro- and anti-angiogenic factors. This delicate, yet dynamic balance between the promotion and inhibition of vascularization provides an abundance of molecular targets for therapeutic intervention.
The search for angiogenic inducers first began after the initial observation that capillary growth was stimulated even when tumors were implanted into avascular regions such as the cornea (1). This evidence of capillary sprouting in areas without or with quiescent vasculature strengthened the hypothesis that tumor cells release diffusible triggers of vascularization.
The first discovered inducer of angiogenesis was basic fibroblast growth factor, known as bFGF or FGF-2 (1). Shortly after its identification, closely related FGF-1, also known as acidic FGF (aFGF), was isolated. Despite a lack of traditional signal sequences for secretion, each growth factor can be released upon exposure to cell stress. One angiogenic protein that is readily secreted from tumor cells, however, is vascular endothelial growth factor (VEGF). Originally identified as vascular permeability factor (VPF) (1), VEGF is a potent promoter of tumor vascularization and is a primary target of anti-angiogenic therapy. Other promoters of angiogenesis include angiopoietins and members of the Ephrin family of ligands (2).
Over the last several decades, a number of pro-angiogenic factors have been identified. Since physiological blood vessel formation is well coordinated and tightly controlled, many hypothesized that tumor angiogenesis is also a complex balance of pro-and anti-angiogenic factors. The existence of endogenous inhibitors of angiogenesis was indeed confirmed when an anti-angiogenic factor was found in the conditioned medium of a nontumorigenic hamster cell line (3). The factor was later purified and shown to be a truncated form of thrombospondin (TSP-1) (1). A secreted glycoprotein that mediates cell–cell interaction, TSP-1 strongly inhibited endothelial cell chemotaxis, as well as vascularization of the cornea. Interestingly, the regulation of TSP-1 levels is dependent on the tumor suppressor protein p53; TSP-1 production is dramatically lower in cells with impaired p53 function (3). This intricate relationship between tumor suppressors and regulators of angiogenesis further emphasizes the pivotal role of vascularization in tumor progression.
Overwhelming evidence suggests that the balance of inducers and inhibitors is critical for promoting vascular quiescence over new capillary formation (1). Not surprisingly, a primary focus within the field of tumor angiogenesis is to prevent the angiogenic switch through the modulation of endogenous factors. In order to increase the efficacy of this therapeutic strategy, however, a deeper understanding of the tumor microenvironment is required.
Current inhibitors and their respective angiogenic targets.
VEGF–VEGFR Pathway
Without question, the most widely studied and well-known target of anti-angiogenesis therapy is VEGF. The mammalian VEGF-related family of angiogenic and lymphangiogenic growth factors consists of five glycoproteins referred to as VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factors 1 and 2 (PGF-1 and PGF-2, respectively) (4, 5). Existing in both soluble and extracellular matrix-bound forms, VEGF promotes endothelial cell growth, migration, and survival in a variety of human cancer types including breast, colorectal, and glioblastoma multiforme (GBM).
Predominantly produced by tumor cells, VEGF ligands exert their pro-angiogenic effects on endothelial cells through the binding and activation of receptor tyrosine kinases known as VEGFR-1, VEGFR-2, and VEGFR-3. Each of the VEGF family ligands has a specific binding affinity for the different receptor tyrosine kinases. VEGF-A binds to both VEGFR-1 and VEGFR-2, while VEGF-B binds exclusively to VEGFR-1. VEGF-C and VEGF-D preferentially bind to VEGFR-3, however, proteolytic processing allows both family members to bind to VEGFR-2 as well (4). Thus, members of the same glycoprotein family are responsible for a diverse range of functions. Since the interaction of VEGF ligands with their respective receptors can trigger the activation of multiple signaling pathways, the VEGF–VEGFR axis has become the most heavily targeted pathway in anti-angiogenesis therapy (4, 5).
Initial approaches to impair VEGF–VEGFR signaling focused on the development of neutralizing antibodies to VEGF ligands (4). Attempts at pharmacological intervention have led to the development of a humanized monoclonal antibody known as bevacizumab (Table I). Recognized for its ability to bind and sequester circulating VEGF-A, bevacizumab was recently approved by the US Food and Drug Administration (FDA) for the clinical treatment of patients with glioma and cancer of the colon, lung, and breast. Additional treatments including sorafenib and sunitinib (inhibitors of VEGF receptor tyrosine kinases, Table I) are also currently approved for cancer therapy (4, 5).
Due to the overwhelming evidence of a pivotal role for VEGF-A in tumor angiogenesis, an array of therapeutic agents has been developed against the VEGF-A signaling pathway. In an initial study of glioma patients who received bevacizumab and a topoisomerase I inhibitor known as irinotecan, 19 out of 29 participants achieved at least a 50% reduction in maximal cross-sectional contrast-enhancing areas on magnetic resonance imaging (MRI) (6). Compared to a radiographic response rate of 5-8% in patients who received standard temozolomide treatment, this result was a marked improvement. Moreover, several large retrospective studies reported response rates of 25-74% and progression-free survival of 32-64% (7-12). These early studies also demonstrated an additional corticosteroid-sparing effect, the majority of patients were able to reduce their corticosteroid doses by 50% or more (6). Since it first entered routine clinical practice in the United States, however, bevacizumab treatment has provided only modest survival benefits overall. Furthermore, although a single infusion has been shown to reduce the density of existing microvasculature (13), bevacizumab primarily targets new blood vessel formation. Thus, bevacizumab monotherapy may lack long-term efficacy because it rarely targets the mature tumor vasculature that is already established at the time of diagnosis. Unfortunately, the molecular mechanism of bevacizumab failure is poorly understood, and the absence of effective post-bevacizumab salvage therapies only complicates treatment management.
Mechanisms of Resistance
Emerging data from clinical and pre-clinical investigations indicate that the aggressive return to tumor growth after initial response to anti-VEGF-VEGFR therapies is due to a variety of drug resistance mechanisms. One mode of resistance to angiogenesis inhibitors is adaptive (evasive) resistance (14). Traditional models of drug resistance focus on the acquisition of genetic mutations within the gene that encodes the drug target. Such mutational alterations lead to reduced inhibition of the intended disease-promoting factor. In the case of evasive resistance, the specific vascular target remains inhibited.
A possible explanation for adaptive resistance, even in the presence of continual drug blockade is that the tumor can activate or up-regulate alternative pro-angiogenic signaling pathways. Evidence for this mechanism in cancer patients was demonstrated in a recent clinical investigation. In participants who received the VEGFR inhibitor cediranib (Table I), there was a temporary response phase followed by relapse. Upon analysis, patient blood levels of bFGF were significantly higher during the relapse phase than in the initial stage of response. Additional observations revealed increased serum levels of stromal cell-derived factor 1α (SDF1α) and elevated presence of circulating endothelial cells at the time of progression on cediranib (6, 15). These findings support the hypothesis that anti-VEGF pathway inhibitors may be more effective when combined with drugs that target other pro-angiogenic factors.
A second mechanism that facilitates evasive resistance is the recruitment of bone marrow-derived cells (BMDCs). BMDCs are recruited in part through increased hypoxia. Hypoxia leading to necrosis is one of the hallmarks of GBM (16), and hypoxic conditions often enhance tumor cell resistance to chemotherapy and ionizing radiation. Interestingly, the inhibition of VEGF-A with bevacizumab produced a substantial increase in hypoxia-inducible factor 1α (HIF1α) and cancer antigen (CA) 9, another hypoxia marker (17). HIF1α was shown to promote GBM neovascularization through the recruitment of pro-angiogenic bone marrow-derived CD45+ myeloid cells and F4/80+ tumor-associated macrophages (14, 18, 19). Tumors with little or no HIF1α expression displayed few BMDCs, and angiogenesis, as well as tumor growth, were significantly impaired. Collectively, these observations suggest that anti-angiogenic therapy induces low oxygen conditions and accelerates blood vessel formation through BMDC recruitment.
Although pro-angiogenic BMDCs are important for progression after anti-VEGF-A therapy, other cell types, such as pericytes, also contribute to evasive resistance. Pericytes, structural support cells that envelop the microvasculature, serve a distinct role of maintaining the integrity and functionality of pre-existing blood vessels. Support for this concept is based on a variety of investigations which showed that increased platelet-derived growth factor (PDGF) signaling enhances the stabilization of tumor blood vessels by recruiting pericytes and promoting pericyte–endothelial cell interactions (6, 20, 21). In light of these observations, combined VEGF–VEGFR and PDGF–PDGFR inhibition would reduce the resistance to antivascular therapy.
A fourth mechanism of adaptive resistance has gained considerable interest over recent years. This form of evasion involves increased invasiveness without the requirement for angiogenesis. Following a pharmaceutical blockade of angiogenesis in an orthotopic mouse model of GBM, glioblastoma cells were able to co-opt the normal vasculature and disseminate deep into the brain (14, 19, 22, 23). This phenotype is referred to as perivascular tumor invasion, and it allows cells to escape oxygen and nutrient deprivation. Consistent with results from animal studies, GBM patients who developed multifocal recurrence after bevacizumab treatment exhibited pro-invasive adaptation as observed by MRI.
Not all modes of resistance are dependent on adaptation. Some tumors possess an intrinsic, pre-existing indifference to anti-angiogenic therapy. In a clinical trial of cediranib, one group of GBM patients exhibited transitory improvements while another subset of patients had no response (6, 15). This differential reaction to the same therapy emphasizes the need for biomarkers that can accurately predict individual efficacy of anti-VEGF-A therapy for individual patients.
Development of Biomarkers for Anti-angiogenic Therapy
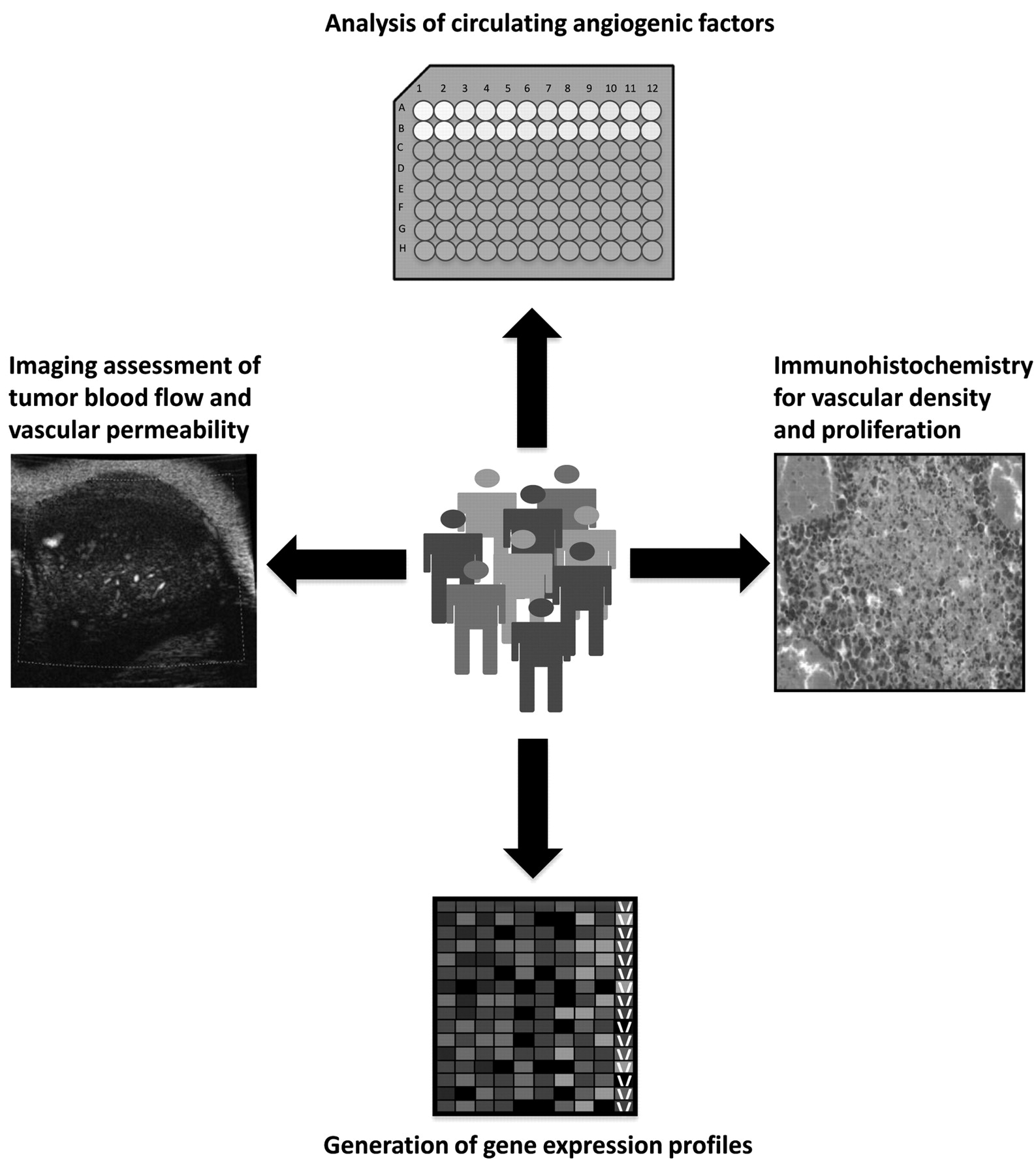
The mechanistic evasion of anti-VEGF-A therapy is further complicated by the lack of indicators or predictive markers for therapeutic efficacy. The contrasting response to anti-angiogenic monotherapy suggests that individual tumors may possess unique angiogenic profiles. These profiles consist of informative parameters such as vascular permeability, blood flow, interstitial fluid pressure, and the up-regulation or down-regulation of key angiogenic factors (24). Deciphering these phenotypic codes on an individualized basis may allow physicians to better predict and monitor a patient's response to various antivascular agents. Although no angiogenic biomarkers have currently been approved for routine clinical use, several measurable parameters may provide prognostic or predictive value in the near future (Figure 1) (25).
One distinctive label of drug activity may be found within the plasma levels of circulating angiogenic factors. In a study of patients with metastatic renal cell carcinoma, plasma VEGF-A and PGF levels were elevated following treatment with SU11248 (Table I), a VEGFR and PDGFR inhibitor (26). Such a response may be indicative of tissue hypoxia or treatment efficacy (25, 26). Furthermore, in a phase I trial of rectal cancer patients, infusion with bevacizumab resulted in a significant increase in plasma VEGF-A and PGF (27). Although further validation is needed, these findings suggest a plausible role for circulating factors in the evaluation of treatment response.
Another promising approach to monitor the effectiveness of anti-angiogenic agents is functional imaging. One commonly used imaging technique is dynamic contrast-enhanced MRI (DCE-MRI). DCE-MRI measures vascular density, tumor blood flow, and vascular permeability by tracking the pharmacokinetics of gadolinium chelate-based contrast agents (25, 28). Recent studies of patients with colon cancer and liver metastasis revealed that DCE-MRI detects response to VEGFR inhibition within two days after treatment (29, 30). In addition, DCE-MRI aids in the selection of high-risk patients with acute myelogenous leukemia for customized anti-angiogenic therapy (28). Due to its simplicity and demonstrated success in a clinical setting, DCE-MRI is rapidly emerging as an accurate means of imaging tumor vascularization. In order for this method to become systematically included for treatment evaluation, however, the acquisition and analysis of DCE-MRI data must undergo standardization across multiple platforms (25, 29).
One method of imaging that exhibits both accuracy and reproducibility is contrast-enhanced ultrasound. Using phospholipid-based microbubbles, both semi-quantitative and quantitative measurements of vessel perfusion can be recorded by means of this imaging technique (25). To date, contrast-enhanced ultrasound imaging has been used to detect vascular changes in a variety of cancer types, including prostate and ovarian (31-33). Moreover, this particular form of ultrasonography is a useful predictor of early sunitinib efficacy in patients with metastatic renal cell carcinoma (34).
Biomarkers for angiogenesis are not solely limited to functional imaging approaches or plasma levels of angiogenic inducers. Mounting evidence indicates that the most favorable assessment of drug response may be found through gene expression profiling (25). Expression analysis of multiple target genes helps create a global description of cellular functions in response to antivascular therapies. Specific gene signatures can then be identified for individual anti-angiogenic drugs, thus allowing physicians to tailor a specific therapeutic regimen to each patient.
Angiogenic biomarkers with diagnostic, prognostic, and predictive value are desperately needed. Combined with standard immunohistological analysis, these measurable parameters could significantly enhance current treatment outcomes. Despite encouraging results from these potential markers, each approach requires further optimization and validation before routine clinical use can be achieved. Until this concept of personalized medicine can be realized, however, a growing number of efforts are focused on improving the potency and selectivity of VEGFR tyrosine kinase inhibitors (TKIs).

Potential biomarkers for anti-angiogenesis therapy. Plausible methods of monitoring clinical response include functional imaging, gene expression arrays, immunohistochemistry of tumor tissue, and analysis of circulating angiogenic factors from patient samples.
Off-target Effects and Toxicity of VEGFR Inhibitors
Although targeting the VEGF family presents a range of challenges, the evidence still suggests that the VEGF–VEGFR axis is the dominant signal transduction pathway in tumor vascularization. Thus, the rationale for using VEGF-A blockade remains quite valid for a variety of cancer types. One potential obstacle to the therapeutic efficacy of VEGFR inhibition, however, is the lack of specificity. FDA-approved TKIs such as sorafenib and sunitinib inhibit VEGFR (Table I), but they also potently block signaling through other targets, including PDGFR, stem cell factor receptor (c-KIT), and colony-stimulating factor 1 receptor (CSF1R) (35). Due to the low selectivity of these multitargeted inhibitors, higher dose administration is required to achieve maximal VEGFR inhibition. As a result, optimal blockade of VEGF-A receptors is often accompanied by increased off-target effects and enhanced toxicity. Adverse events associated with reduced target selectivity include hand–foot skin reactions, fatigue, vomiting, diarrhea, hypertension, cardiac ischemia, and thyroid dysfunction (35). Consequently, the combined use of TKIs with conventional chemotherapeutic drugs is also severely limited.
Second-generation VEGFR TKIs
To circumvent the problems associated with off-target toxicities, a new initiative is underway to develop second-generation VEGFR TKIs that exhibit extreme potency and elevated selectivity. One of the most promising candidates is a pan-VEGFR antagonist known as tivozanib (Table I). Demonstrating picomolar potency to VEGFR-1, VEGFR-2, and VEGFR-3, tivozanib significantly increased progression-free survival from 6.2 months to 12.1 months in patients with renal cell carcinoma (35). Furthermore, it is the first TKI to be safely combined with an inhibitor of mammalian target of rapamycin (mTOR) and is currently involved in phase I studies with other therapeutic agents for both metastatic breast cancer and advanced colorectal cancer. Tivozanib is not alone in this subset of novel selective TKIs. Axitinib (Table I), a small-molecule inhibitor of all known VEGFRs, also demonstrates increased efficacy as both monotherapy and in combination with chemotherapy. Further clinical evaluation is being performed to assess the effects of axitinib in lung cancer, metastatic breast cancer, pancreatic cancer, and advanced gastric cancer (35). Overall, collective data generated from clinical studies indicate that second-generation TKIs are generally associated with lower off-target toxicities, and are more potent and less toxic than traditional inhibitors of receptor tyrosine kinases.
Protein Kinase D Family as Anti-angiogenic Targets
Since the tolerability of novel TKIs is still unknown for multiple tumor types, another potential approach to controlling the angiogenic process is to target the VEGF–VEGFR pathway through indirect mechanisms of action. In this modified approach to vascular inhibition, therapeutic strategies are designed to block the downstream effectors of VEGF-A-induced angiogenic signaling. Recent investigation of tumor blood vessel formation revealed a key role for protein kinase D1 (PKD1) in VEGF-A signaling (36).
A novel serine/threonine protein kinase, PKD1 mediates VEGFR-2-stimulated endothelial cell proliferation through the activation of extracellular signal-regulated kinases 1 and 2 (ERK 1/2) (37). Moreover, the subcutaneous implantation of matrigel plugs in vivo showed that functional PKD1 activity was necessary for VEGF-A-induced angiogenesis. The ability of PKD1 to promote angiogenesis is due in part to class IIa histone deacetylase (HDAC) activity. Important for chromatin modifications and repression of gene expression, HDAC5 and HDAC7 enzymes are direct targets of PKD1-dependent phosphorylation in endothelial cells stimulated with VEGF-A. Upon phosphorylation of Ser259/498 in HDAC5 and Ser178, Ser344, and Ser479 in HDAC7, HDAC translocation from the nucleus to the cytoplasm promotes the expression of myocyte enhancer factor-2 (MEF2)-dependent genes (37). Through an unidentified mechanism, the PKD1–HDAC pathway eventually leads to angiogenic gene expression, endothelial cell migration, tubule formation, and microvascular sprouting.
As a frequently up-regulated isoform in pancreatic and prostate cancer, PKD1 has understandably become an attractive target for chemical inhibition. Perhaps the most promising PKD inhibitor is CRT0066101 (Table I) (36). A pan-inhibitor of PKD1, PKD2, and PKD3, CRT0066101 is orally available and substantially suppresses the growth of pancreatic tumors in an orthotopic mouse model. Unfortunately, information regarding the three-dimensional structure of PKD is incomplete. Therefore, the present lack of structure-based drug design hinders the optimization of current anti-PKD compounds.
Inhibitors of Rho GTPase Signaling
Other downstream mediators of VEGF–VEGFR signaling are also generating interest as proponents of tumor angiogenesis. One particular signaling cascade, the Ras homolog (Rho) GTPase pathway, has recently been implicated in several phases of angiogenesis, such as vascular permeability, endothelial cell migration, proliferation, and lumen formation. Functioning as molecular gatekeepers, this subfamily of the Ras superfamily of small GTPases is activated by VEGF-A binding to VEGFR-2 in endothelial cells (38).
This ligand-receptor interaction initiates the recruitment of proteins such as c-Src or phospholipase C beta 3 (PLCβ3) to the phosphorylated tyrosine residues of the VEGF-A receptors. Following this initial recruitment, Rho GTPases such as Ras-related C3 botulinum toxin substrate 1 (Rac1), RhoA, and cell division control protein 42 homolog (Cdc42) become active and subsequently promote tumor vascularization through destabilization of endothelial barrier integrity, enhanced migration, proliferation, and tubule formation (38).
Attempts to disrupt Rho GTPase signal transduction frequently involve the use of Rho-associated coiled-coil-forming kinase (ROCK) inhibitors. One such inhibitor, HA1077 (fasudil) (Table I), can effectively block migration, cellular viability, and tubule formation in VEGF-A-stimulated vascular endothelial cells. Interestingly, treatment with fasudil attenuates the anchorage-dependent growth of breast cancer cells and significantly reduces tumor burden in an experimental model of murine lung metastasis (38).
Another compound, NSC23766 (Table I), is a small-molecule inhibitor of Rac1. Similar to ROCK inhibitors, NSC23766 attenuates cell proliferation, as well as tumor growth. In contrast to the effects of pharmacological inhibition of Rac1, however, depletion of Rac1 in the tumor endothelium of adult wild-type mice had no effect on angiogenesis and revealed a complete lack of tumor growth suppression (38). In order to reconcile such conflicting outcomes, more information is needed regarding the regulation and downstream signaling of Rho GTPases. Unraveling the intricate relationships between different members of this signaling family may uncover the necessary combinations of druggable targets that will be most effective for clinical use.
Novel Molecular Targets for Anti-angiogenic Therapy
For years, the VEGF-A signaling pathway has been the primary target of vascular inhibition. Due to the emerging complications of resistance, however, efforts to discover new molecular targets have increased.
Cytosolic phospholipase A2. We have previously described the role of cytosolic phospholipase A2 (cPLA2) in radiation-induced signal transduction in human lung cancer and ovarian carcinoma (39-41). Following the observation that cPLA2 promotes the survival of vascular endothelium, we have also identified this cytoplasmic enzyme as a fundamental component of tumor angiogenesis (42). In cPLA2α-deficient mice, a syngeneic glioblastoma cell line (GL261) failed to form tumors even 2 months after injection. By contrast, their cPLA2α-wild type counterparts displayed a tumor take rate of 100%. In similar experiments, Lewis lung carcinoma (LLC) cells did form tumors in cPLA2α-deficient mice; however, they were dramatically smaller than tumors in cPLA2α wild-type mice. Immunohistochemical examination revealed reduced blood vessel formation and increased areas of necrosis in tumors from cPLA2α−/− mice compared to wild-type mice, thus implicating cPLA2 as an important factor for tumor formation, growth and maintenance (42).
Tumor vascularization requires not only capillary formation, but also the maturation of endothelial-lined blood vessels. This process of vessel maturation is dependent on the functions of cells known as pericytes. Previously regarded as inactive scaffolding components, pericytes are now recognized for their ability to coordinate intercellular signaling with endothelial cells and other elements of the blood vessel wall to prevent leakage. As necessary components in vessel stabilization, pericytes maintain vessel integrity and aid in the assembly of extracellular matrix (ECM) components. By using immunohistochemical analysis of blood vessels from LLC tumors formed in cPLA2α-deficient and wild-type mice, we demonstrated a critical role of cPLA2α for the presence of both mature and immature pericytes (42). These marked differences in pericyte coverage suggest that in addition to its role in endothelial cell function, cPLA2 may also be responsible for pericyte recruitment and required vessel maturation. Such findings implicate a novel pro-angiogenic role for cPLA2 in the tumor microenvironment. Moreover, we also demonstrated that cPLA2α-dependent production of bioactive lipid mediators lysophosphatidylcholine (LPC), arachidonic acid (AA), and lysophosphatidic acid (LPA) contribute to the promotion of vascular endothelial cell functions (42). While AA and LPA are known to regulate endothelial cell migration (43-46), the LPC pathway is a newly identified significant player in this process. In addition, the most pronounced increase in cellular proliferation was observed in cPLA2α−/− cells treated with a combination of LPC and LPA (42). These data demonstrate a key role for cPLA2 in endothelial cell proliferation and indicate that the lysophospholipids, LPC and LPA, may serve as effectors for this stage of angiogenesis.
Due to its functional responsibility in inflammation, radiation signaling, and angiogenesis, cPLA2 has become an attractive target for chemical inhibition. Two of the most commonly used cPLA2 inhibitors are arachidonyltrifluoromethyl ketone (AACOCF3) and methyl arachidonyl fluorophosphonate (MAFP) (Table I) (41). AACOCF3 is a potent and cell-permeable trifluoromethyl ketone analog of arachidonic acid. Nuclear magnetic resonance studies have shown that the carbon chain of AACOCF3 binds in a hydrophobic pocket of cPLA2 and the carbonyl group of AACOCF3 forms a covalent bond with serine 228 in the enzyme active site (47). This cPLA2 inhibitor has been used to study the role of cPLA2 in platelet aggregation, inflammation-associated apoptosis, and the radiosensitivity of vascular endothelial cells (41). Combined with radiation, AACOCF3 was also demonstrated to significantly inhibit tumor growth and tumor vascularity in mouse models of lung and ovarian cancer (39, 40). Similar to AACOCF3, MAFP is also a powerful inhibitory agent, however, this irreversible drug inhibits both the calcium-dependent and calcium-independent (iPLA2) forms of the enzyme (47). Other agents including pyrrolidine-based inhibitors have also been used extensively to block PLA2 enzymatic activity. Nevertheless, like other pyrrolidine-containing compounds, this class of drugs is non-specific and attenuates the activity of cPLA2-γ and iPLA2-β (47). Recent attempts to improve the specificity of these compounds have unveiled new indole-based candidates for cPLA2-targeted therapy (48). Based on promising preliminary results, our laboratory synthesized a compound known as 4-[2-[5-chloro-1-(diphenylmethyl)-2-methyl-1H-indol-3-yl]-ethoxy]benzoic acid (CDIBA) (Table I) (42). Shown to potently target cPLA2α and substantially attenuate arachidonic acid release in a wide variety of enzymatic and cell-based assays, CDIBA treatment inhibited capillary tubule formation, migration, and cellular proliferation in tumor vascular endothelial cells (42). Furthermore, in heterotopic glioblastoma and lung cancer tumor models, mice treated with CDIBA exhibited delayed tumor growth and reduced tumor volume (42). Accordingly, pharmaceutical companies are now focused on the continual optimization of these novel cPLA2 inhibitors for clinical use.
Autotaxin and LPA signaling. Advances in anti-angiogenic therapy are not solely dependent on the discovery of endothelial-associated targets, however. Indeed, as in the case of VEGF-A, tumor cells also secrete soluble inducers of angiogenesis. An excellent example of this paracrine effect can be found in the previously mentioned lipid second messenger, LPA. Shown to stimulate cell proliferation, migration, and survival, LPA has been implicated in the progression of many tumor types including lung cancer, hepatocellular carcinoma, and epithelial ovarian cancer (46, 49). LPA signaling is primarily mediated through classic G protein-coupled receptors that belong to the endothelial differentiation gene family (LPA1, LPA2 and LPA3) LPA can also exert its role through other receptors, such as LPA4, LPA5, probable G-protein coupled receptor 87 (GPR87) and P2Y purinergic receptor 5 (P2Y5) (50). There are two major pathways of LPA production: i) phosphatidic acid generated by phospholipase D or diacylglycerol kinase is subsequently converted to LPA by cPLA2; ii) lysophospholipids generated by cPLA2 (such as LPC) are subsequently converted to LPA by lysophospholipase D, also known as autotaxin (ATX) (50-54). Unlike other members of the ectonucleotide pyrophosphatase and phosphodiesterase (NPP) family of enzymes, ATX possesses robust lysophospholipase D activity (46, 52). Initially purified from melanoma cells as a potent chemoattractant (55), expression of this 103 kDa secreted protein is up-regulated in a variety of malignancies and has been shown to stimulate cell proliferation and enhance tumor invasion and metastasis (56).
While ATX can be produced by endothelium, the majority of ATX is generated and secreted by a variety of tumor cells. As the primary enzyme involved in the production of LPA, ATX has recently sparked interest for its potential role in the development and progression of ovarian cancer. As the fifth leading cause of death in American women, ovarian tumors are known to produce larger quantities of LPA than nonmalignant cells (57). Due to the high substrate levels of LPC found in peritoneal fluids from patients with ovarian cancer, the observed increase in LPA is attributed to elevated ATX activity (40, 53). Moreover, expression of LPA receptors was found to determine tumorigenicity and aggressiveness of ovarian cancer cells. Our laboratory and others have recently demonstrated that ATX and LPA signaling may contribute to the resistance of ovarian carcinoma to cytotoxic therapies such as cisplatin and ionizing radiation (40). Based on these results, it remains conceivable that LPA generated from LPC hydrolysis could bind to LPA receptors on vascular endothelium, as well as those found on tumor cells.
Correspondingly, a cooperation of both VEGF-A and ATX was discovered in the regulation of endothelial cell migration (46). Knockdown of ATX expression prevented endothelial cell migration in response to stimulation with LPC, LPA, and VEGF-A. Moreover, the genetic silencing of ATX resulted in a concomitant reduction in the mRNA levels of LPA receptors (46). Taken together, these data suggest that ATX regulates the expression of LPA receptors that are necessary for VEGF-A- and lysophospholipid-induced angiogenesis. Thus, pharmacological inhibition of both ATX and LPA may serve as an effective method to reduce tumor vascularization.
Since ATX is a member of the alkaline phosphatase superfamily of metalloenzymes, initial medicinal chemistry efforts focused on metal chelaters as ATX inhibitors (58). The chelaters reduce the metal-ion stimulation of ATX activity by competing with active site histidine and aspartic acid residues for divalent metal ions (53, 58, 59). Not surprisingly, metal–ion chelation is considered a relatively insensitive and non-specific method of ATX inhibition (58). The library of ATX inhibitors has since expanded to include both non-lipid small-molecule inhibitors, as well as analogs of bioactive lipids. Both categories of drugs are promising in vitro, although the non-lipid ATX inhibitors are more compliant with the characteristic parameters often found for orally bioavailable drugs (58, 60). Furthermore, these non-lipid compounds exhibit enhanced specificity for ATX without affecting other members of the NPP family (58). Despite the identification of new inhibitory agents, the lack of information regarding the three-dimensional structure of ATX is impeding drug discovery (61). Until the structural details of the enzyme are publicly disclosed, current medicinal chemistry efforts are focused on other ATX-associated targets, such as LPA and the LPA receptors. One of the most common approaches to disrupting LPA signaling is to use LPA derivatives as selective receptor antagonists (62). Many of these individual derivatives can exert a combined antagonistic effect against more than one LPA receptor. A primary example of this can be found in the α–bromomethylene phosphonate analog of LPA known as BrP-LPA (Table I). As a pan-antagonist of LPA1-4, BrP-LPA has been shown to significantly reduce migration, invasion, vascularity and tumor volume in mouse models of breast cancer and non-small cell lung cancer (62-66). Moreover, this potent and efficacious inhibitor also blocks over 98% of ATX activity at micromolar concentrations (65). Such results suggest that the use of multitarget antagonists may provide the best strategy for the abrogation of ATX-LPA signal transduction.
Enhancer of Zeste Homolog 2. The combined approach to antivascular therapy is becoming increasingly attractive as the tumor-endothelial cell interaction is deciphered. For instance, a paracrine relationship between VEGF-A and the enhancer of Zeste homolog 2 (EZH2) was recently identified (67). EZH2 is a member of the polycomb-group (PcG) proteins and has intrinsic histone methyl transferase activity. Histone methylation is a common method of epigenetic gene regulation and is typically associated with transcriptional repression. Armed with this ability to inhibit transcription, EZH2 is frequently implicated in tumor progression and metastatic disease. A recent investigation revealed that VEGF secreted from human epithelial ovarian cancer cells was able to directly increase EZH2 mRNA levels in vascular endothelial cells (67). Elevated tumoral and endothelial EZH2 was also observed in more than 60% of available epithelial ovarian cancer samples. Furthermore, heightened levels of EZH2 were associated with high-grade disease and were predictive of poor overall survival. In a study to determine the mechanism behind such a dismal clinical outcome, Lu and colleagues investigated the relationship between EZH2 and the secreted protein, vasohibin1 (VASH1). Induced by VEGF-A stimulation, VASH1 is a newly identified negative regulator of angiogenesis. Interestingly, increased EZH2 resulted in the methylation and subsequent inactivation of VASH1 (67). Considering the complications of intrinsic or acquired resistance to anti-VEGF-A monotherapy, a combinatorial strategy that focuses on vascular and tumor-specific targets may provide the greatest efficacy.
Despite growing evidence of the relationship between EZH2 and tumorigenesis, there are currently no clinically available therapies that directly target histone methylation (68). Some experimental studies on the inhibition of EZH2 activity have been performed, however. Using a carbocyclic adenosine analog known as 3-deazaneplanocin (DZNep) (Table I), several groups have demonstrated depletion of EZH2 levels and reduced proliferation in breast cancer and prostate cancer cells (68, 69). Futhermore, treatment with DZNep induced apoptosis in acute myeloid leukemia (AML) cells and significantly prolonged the survival of mice implanted with AML cells (70). Similar to other non-specific inhibitors, DZNep affects targets other than EZH2 (68, 70, 71). Although EZH2 is the catalytic subunit of the polycomb repressive complex 2, it is accompanied by other components including suppressor of zeste 12 homolog, embryonic ectoderm development protein, and yin yang 1. Consequently, treatment with DZNep results in the depletion of each of these PRC2 complex proteins and blocks methylation of the associated histone H3 lysine 27 (68, 70, 71). Therefore, DZNep may interfere with normal physiological processes that require methyl transfer.
Cyclin-dependent Kinases. Cyclin-dependent kinases (CDKs) have long been recognized for their involvement in the regulation of cell cycle transitions and cellular proliferation. As members of the serine-threonine kinase family, CDKs bind to regulatory proteins called cyclins and phosphorylate protein substrates on serine and threonine amino acid residues. Given the importance of cell cycle management in the prevention of uncontrolled cell growth, studies that shed light on the function of CDKs in tumorigenesis have recently gained momentum. A number of small-molecule inhibitors have been developed to alter the CDK deregulation that is frequently observed in human cancer (72-74). Success with one of the earliest CDK inhibitors, olomoucine, led to the widespread search for more specific compounds that would preclude aberrant CDK activity in tumors. To date, multiple CDK inhibitors have demonstrated antiproliferative effects in cultured and xenografted myeloma, leukemia, colon cancer, lung cancer, and breast cancer cells (72-74). Recently, the CDK inhibitor roscovitine (Table I), was shown to arrest human estrogen receptor alpha (ER-α) positive MCF-7 breast cancer cells in the G2 phase of the cell cycle and induce p53-dependent apoptosis (75). Based on its anticancer activity both in vitro and in vivo, roscovitine is being evaluated in phase II clinical trials for the treatment of non-small cell lung cancer and nasopharyngeal cancer (72-74). Aside from its ability to impede tumor cell division, anti-angiogenic properties have also been discovered for this CDK inhibitor. Surprisingly, only a few prior reports have denoted a role for CDKs in tumor angiogenesis. To understand the molecular basis of these cell cycle and transcriptional regulators in tumor blood vessel formation, Liebl et al. assessed the effects of CDK inhibition in human umbilical vein endothelial cells (73, 74). In response to treatment with roscovitine, endothelial migration and tubule formation was significantly reduced. Furthermore, the chemical inhibition of CDKs greatly impaired endothelial cell sprouting from mouse aortic rings and abolished VEGF-A-induced vessel formation in the chorioallantoic membrane assay (73, 74). While roscovitine does not selectively inhibit one specific CDK, the knockdown of CDK5 revealed that roscovitine might exert its antivascular properties through a CDK5-dependent pathway (73, 74). Other CDKs, such as CDK4, have also been reported as plausible contributors to tumor vascularization. In a murine model of intestinal tumors, constitutive activation of CDK4 was shown to enhance tumor blood vessel formation and increase the expression of E2F target proteins involved in angiogenesis and proliferation (72, 76). Taken together, these findings suggest that the pharmacological inhibition of CDKs, either alone or in combination, may provide a novel method of vascular destruction (72-74).
Conclusion
Current attempts to disrupt the complex process of tumor blood vessel formation are predominantly focused on the VEGF–VEGFR signaling pathway. Although clinically proven to inhibit VEGF-A and its receptors, these pharmacologic agents are selective, but not specific. Consequently, many of the approved inhibitors also impair other molecular targets, thus leading to increased toxicity. To reduce toxicity complications and augment the destruction of the tumor vascular network, an active search for new inhibitory agents has begun. In recent years, the emergence of several VEGF–VEGFR angiogenesis inhibitors has enhanced the clinical outcome of patients for multiple types of tumor. It is important to note, however, that many of these pharmacologic agents resulted in transitory improvements followed by increased tumor resistance and metastasis. The observed resistance may be partially explained by the complex network of signal transduction that constitutes the angiogenic process. The frequent interconnectivity of these signaling pathways often results in redundancy during the formation of tumor blood vessels. As a result, when one pro-angiogenic target is inhibited, other molecules can be activated so that the requirement for vascularization is once again fulfilled. Thus, the most effective therapeutic strategy may be to combine conventional treatment regimens with therapies that target multiple angiogenic pathways.
- Received November 9, 2011.
- Revision received December 2, 2011.
- Accepted December 5, 2011.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
Jump to section
- Article
- Abstract
- VEGF–VEGFR Pathway
- Mechanisms of Resistance
- Development of Biomarkers for Anti-angiogenic Therapy
- Off-target Effects and Toxicity of VEGFR Inhibitors
- Second-generation VEGFR TKIs
- Protein Kinase D Family as Anti-angiogenic Targets
- Inhibitors of Rho GTPase Signaling
- Novel Molecular Targets for Anti-angiogenic Therapy
- Conclusion
- References
- Figures & Data
- Info & Metrics