


Abstract
Background: The synthetic retinoid fenretinide (N-(4-hydroxyphenyl)retinamide, 4-HPR) has shown promising anticancer activity in preclinical studies, but its limited oral bioavailability has hindered clinical assessment. A novel lipid matrix, Lym-X-Sorb (LXS), was evaluated to improve fenretinide bioavailability and attain higher plasma concentrations. Patients and Methods: Adults with refractory malignancies were administered fenretinide/LXS oral powder in 2 divided doses over 24 h for 7 consecutive days every 21 days in a standard phase I dose-escalation study with pharmacokinetic analysis. Results: The principal toxicities observed were diarrhea, reversible night blindness, and allergic reaction. The maximum tolerated dose regimens were 1,000 mg/m2/day divided into 2 daily doses for 7 days, every 21 days, and 800 mg/m2/day divided into 3 daily doses for 7 consecutive days, every 21 days. Conclusion: Better fenretinide formulations are needed to improve adult patient acceptability and compliance and to achieve the consistent systemic exposures associated with activity in preclinical models.
Fenretinide (N-(4-hydroxyphenyl)retinamide, 4-HPR, NSC 374551) is a synthetic retinoid with cytotoxic and antitumor activity in multiple cell lines and preclinical models (1-5). The mechanism of fenretinide cytotoxicity is incompletely defined. Evidence includes support for an increase in reactive oxygen species and dihydroceramide levels (via stimulation of de novo synthesis) in a retinoid receptor–independent manner, and cytotoxicity in a p53-independent manner in tumor cell lines, in vitro (2).
Based on promising preclinical activity, an oral corn-oil capsule was evaluated in clinical trials at low doses (100-400 mg/day, 1-3 μM plasma levels) as a chemopreventive agent, and at higher doses (1,800-4,200 mg/day, 7-10 μM plasma levels) for the treatment of advanced solid tumors (6-9). Treatment was generally well tolerated, with reversible nyctalopia (decreased dark adaptation vision) due to depletion of plasma retinol levels, dry skin, and rash observed at lower doses, and reversible hepatic dysfunction, hypertriglyceridemia, idiosyncratic pediatric pseudotumor cerebri, nausea, and mild thrombocytopenia observed at higher doses. A wide interpatient variability in attained plasma drug levels suggested inconsistent absorption.
A novel lipid matrix, termed Lym-X-Sorb (LXS), was used to formulate fenretinide to improve the bioavailability of this poorly water-soluble agent and to attain higher plasma concentrations (10). With the LXS system, a lipophilic drug, such as fenretinide, is encapsulated in a lipid matrix for oral delivery (to resemble a chylomicron) and absorbed through the proximal intestine into the lymphatic system, entering the systemic circulation via the thoracic duct. The empty LXS lipid matrix has been successfully employed to increase the delivery of fat-soluble vitamins and as a lipid nutritional supplement in patients with cystic fibrosis (11). For the present study, fenretinide in LXS matrix was blended with wheat flour and sugar to form a free-flowing powder (fenretinide/LXS oral powder) that could be delivered in any non-milk fat-containing food product such as oatmeal or applesauce. In a single-dose pilot study in mice comparing the fenretinide/LXS oral powder to the contents of corn oil-containing fenretinide capsules, administration of fenretinide/LXS oral powder produced drug plasma levels of ~10 to 20 μM, which were four- to six-fold higher than those achieved with the capsules (10).
Based on these data, a phase I dose-escalation study was conducted to determine the safety, toxicity, maximum tolerated dose (MTD), and pharmacokinetics of fenretinide/LXS oral powder given orally for 7 consecutive days, every 21 days, in adult patients with recurrent and/or resistant solid tumors or lymphomas.
Patients and Methods
Eligibility criteria. Patients (age ≥18 years) were eligible if they had pathologically confirmed metastatic or unresectable malignancy for which there was no acceptable standard therapy; an Eastern Cooperative Oncology Group performance status ≤2; and adequate organ and marrow function defined as absolute neutrophil count ≥1,500/μl, platelets ≥100,000/μl, total bilirubin ≤1.5 × the upper limit of normal (ULN), aspartate aminotransferase and/or alanine aminotransferase <2.5 × ULN, and creatinine <1.5 × ULN.
Prior anticancer therapy must have been completed at least 4 weeks before starting the study drug. Patients were excluded if they had an uncontrolled intercurrent illness, untreated brain metastases, were pregnant or lactating, or had allergy to wheat gluten. Patients with a history of pancreatitis (abdominal pain with elevated amylase or lipase) were excluded due to the potential to worsen pancreatitis from an elevation in plasma lipids.
The protocol design and conduct followed all applicable regulations, guidances, and local policies; (ClinicalTrials.gov Identifier: NCT00589381).
Trial design. This was an open-label, single-arm phase I study of fenretinide/LXS oral powder in patients with advanced malignancies. Fenretinide/LXS oral powder tested in this trial was produced with assistance from the National Cancer Institute's (NCI) Rapid Access to Intervention Development program and supplied by the Texas Tech University Health Sciences Center (Lubbock, TX, USA) under an agreement with the Division of Cancer Treatment and Diagnosis, NCI. Fenretinide was prepared by mixing the fenretinide/LXS oral powder in 2 to 10 oz. of non-milk fat-containing food product such as oatmeal, applesauce, or apple juice. The fenretinide/LXS oral powder preparation contained 2.2% fenretinide by weight, i.e. 4 g of the powder contained 88 mg fenretinide. The total daily dose was calculated based on the patient's body surface area and then divided for administration in 2 equally divided doses over a 24-h period (approximately 12 h apart). The patients were given a 10-cc scoop and precise instructions on how many scoops to use per dose and how to prepare the mixture. Drug was administered for 7 consecutive days of a 21-day cycle. Once the MTD was established for the twice-a-day schedule, the total dose was divided into 3 doses to be administered approximately 8 h apart over a 24-h period, and the MTD was established for that schedule. The starting dose was 1,000 mg of fenretinide/m2/day, which was 45% of the dose eventually established as tolerable in a study of this preparation in relapsed pediatric neuroblastoma (NANT 2004-04 study) (7). A standard phase I dose-escalation design was employed, whereby groups of three to six patients were treated at each dose level. Doses were increased approximately 30% between dose levels; higher dose levels were not opened to accrual until the last patient in the previous cohort had completed two cycles. Intrapatient dose escalation was allowed.
Adverse events were graded according to NCI Common Toxicity Criteria version 3.0 (http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_30). Dose-limiting toxicity (DLT) was defined as an adverse event that occurred in the first cycle, was felt to be related to the study drug, and fulfilled one of the following criteria: grade 4 hematological toxicity (except grade 4 neutropenia lasting <5 days without fever); grade 3 or greater non-hematologic toxicity (except reversible nyctalopia, headache not due to pseudotumor cerebri, and nausea, vomiting, and diarrhea that responded to supportive measures); any evidence of pancreatitis; or toxicity delaying treatment for more than 7 days. All visual toxicities were carefully evaluated and discussed with the principal investigator before proceeding with further drug administration. Significant loss of visual acuity required immediate evaluation by an ophthalmologist, and was defined as loss of three lines of vision by Snellen visual acuity testing, and/or 5 feet in linear Allen testing. Patients could receive further therapy on study with a dose reduction if symptoms completely resolved. Diarrhea was treated with loperamide 2 mg p.o. every 2 h while awake and every 4 h while asleep until the patient was free of diarrhea for 12 h. This regimen could be repeated for each diarrheal episode. The occurrence of liquid stools after a 12-h diarrhea-free period was considered a new episode. Diarrhea that did not resolve to at least a grade 2 within 24 h of starting the anti-diarrheal treatment was considered refractory. The following cycle was delayed if serum triglyceride values were 300 mg/dl or higher, and patients were started on medical therapy with lipid-lowering agents. If, despite medical management, serum triglycerides were 300 mg/dl or higher at the start of the next cycle, the dose was reduced.
Patients were instructed to maintain a study diary recording the daily intake of fenretinide/LXS oral powder, including the time the dose was taken, the number of scoops consumed, and whether or not any doses were missed. Patients also recorded any new symptoms or changes in concomitant medications.
Safety and efficacy evaluations. History, physical examination, ophthalmology evaluation, echocardiogram, complete blood counts (CBC) with differential, serum chemistries, serum triglyceride levels, urine pregnancy test, and radiological imaging of the head, chest, abdomen, and pelvis were performed at baseline. History, physical examination, serum chemistries, and serum triglyceride measurements were performed on days 1 and 7 of each cycle; CBC with differential was performed weekly throughout the cycles. Ophthalmology examinations were repeated if patients developed symptoms. Radiographic evaluation was performed at baseline and every two cycles to assess for tumor response based on the Response Evaluation Criteria in Solid Tumors version 1.0 (12).
Pharmacokinetics of fenretinide. Blood samples (4 ml in heparin-containing tube) were drawn during cycles 1, 2, and 6 on days 1 and 7, prior to drug administration and at 2, 4, 6, and 8 h after the morning dose. Samples were protected from light; plasma was separated by low-speed centrifugation (1,500 rpm for 10 min) and then transferred to a labeled, foil-wrapped polypropylene tube, and frozen immediately at −70°C.
Plasma concentrations of 4-HPR and its main metabolite N-(4-methoxyphenyl)retinamide (4-MPR) were quantified in plasma using an high-performance liquid chromatography (HPLC) assay as previously described (13) with the following modifications: HPLC equipment was an Agilent 1200 system (Agilent Technologies, Palo Alto, CA, USA); column was an Agilent Zorbax Eclipse reverse-phase C18 150×4.6 mm, 5 μm (Agilent Technologies); gradient elution with 0.01 M ammonium acetate in water (W) and methanol (M) was used at a flow rate of 1 ml/min and the gradient (given as W/M, v/v) was 50/50 until minute 2, linear increase to 5/95 by minute 15, 5/95 until minute 20, linear decrease to 50/50 by minute 23, and 50/50 until minute 25. Calibration curve concentrations ranged from 0.31 to 40 μg/ml for both 4-HPR and 4-MPR; injection volume into the HPLC was 30 μl; and 100 μl of each plasma or tissue sample was used for extraction and analysis. Data were acquired and integrated by ChemStation 3D system software (Agilent Technologies). 4-HPR, 4-MPR, and internal standard N-(4-ethoxyphenyl)retinamide (4-EPR) were supplied by the Development Therapeutics Program, NCI (Bethesda, MD, USA). Ammonium acetate, HPLC-grade methanol, and reagent-grade formic acid were purchased from Sigma-Aldrich (St Louis, MO, USA).
Results
Twenty patients were enrolled. Patient characteristics are detailed in Table I. A total of 68 cycles of therapy were administered; one patient received 24 cycles of therapy. The first three patients were enrolled into Dose Level 1, 1,000 mg/m2/day administered in two divided doses. There were no DLTs, and the dose was escalated to 1300 mg/m2/day (Dose Level 2) for the subsequent cohort. Out of the three patients on Dose Level 2, one patient developed a DLT (refractory grade 3 diarrhea) and another had a DLT consisting of grade 3 allergic reaction to the oral fenretinide. The patient with the allergic reaction presented with a diffuse erythematous pruritic rash covering the entire body within 24 h of the first dose of the study drug. Skin biopsy revealed perivascular infiltrates with eosinophilia, consistent with a drug reaction. The patient was subsequently treated with parenteral steroids and antihistamines, which resolved the reaction. This patient had a history of similar allergic reactions to platinum-based chemotherapy and cephalosporins and was taken off the study due to toxicity. Due to the occurrence of two DLTs at Dose Level 2, dose escalation was stopped, and three additional patients were enrolled on Dose Level 1. One of these three patients experienced a DLT (refractory grade 3 diarrhea). Because 1 out of 6 total patients developed a DLT at this dose level, 1,000 mg/m2/day divided into 2 daily doses for 7 days, every 21 days, was declared the MTD.
Because the principal DLT was diarrhea, considered secondary to the formulation and the amount of fat content per dose administered, an alternate schedule of the total daily dose divided in three doses every 8 h was subsequently evaluated following a protocol amendment. Of the four patients enrolled into the 1,000 mg/m2/day dose level of the new schedule, one patient developed refractory grade 3 diarrhea; this patient's dose was reduced to 800 mg/m2/day, and further therapy was well tolerated. Another patient developed grade 4 thrombocytopenia and a grade 2 allergic reaction to the oral fenretinide. Since two DLTs occurred at 1,000 mg/m2/day on the 3-times-daily schedule, subsequent patients were enrolled onto a dose level of 800 mg/m2/day, divided into 3 doses per day, for 7 days, every 21 days. Six patients received this dose and schedule, and no DLTs were observed. Therefore, 800 mg/m2/day, divided into 3 doses over a 24-h period, administered days 1 through 7, on a 21-day cycle, was determined to be the MTD for this schedule.
Patient characteristics.
Seven of the 20 study patients refused further therapy due to the gastrointestinal (GI) side-effects or the taste of the study drug. Five of these 7 patients had grade 1 or grade 2 GI side-effects controlled by concomitant medications. The principal reason for patients withdrawing from the study was the taste and texture of the study medication.
One patient with cutaneous T-cell lymphoma had disease stabilization with improvement in pruritus, and resolution of skin infections, with improvement in lower extremity ulceration felt to be secondary to ischemic changes. This patient tolerated oral fenretinide well and received a total of 24 cycles of treatment with minimal GI symptoms. He developed reversible nyctalopia during each cycle, lasting up to 10 days per cycle. A list of adverse events for the trial as a whole can be found in Table II.
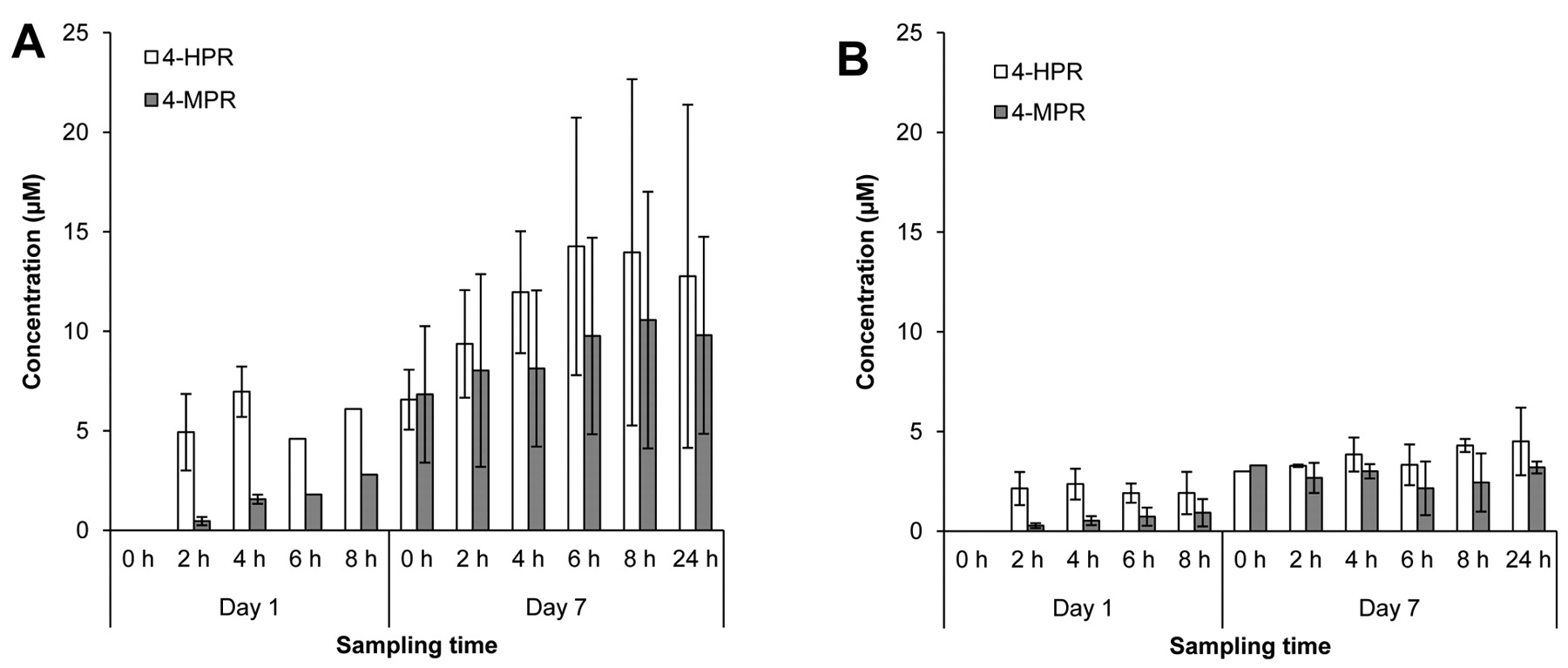
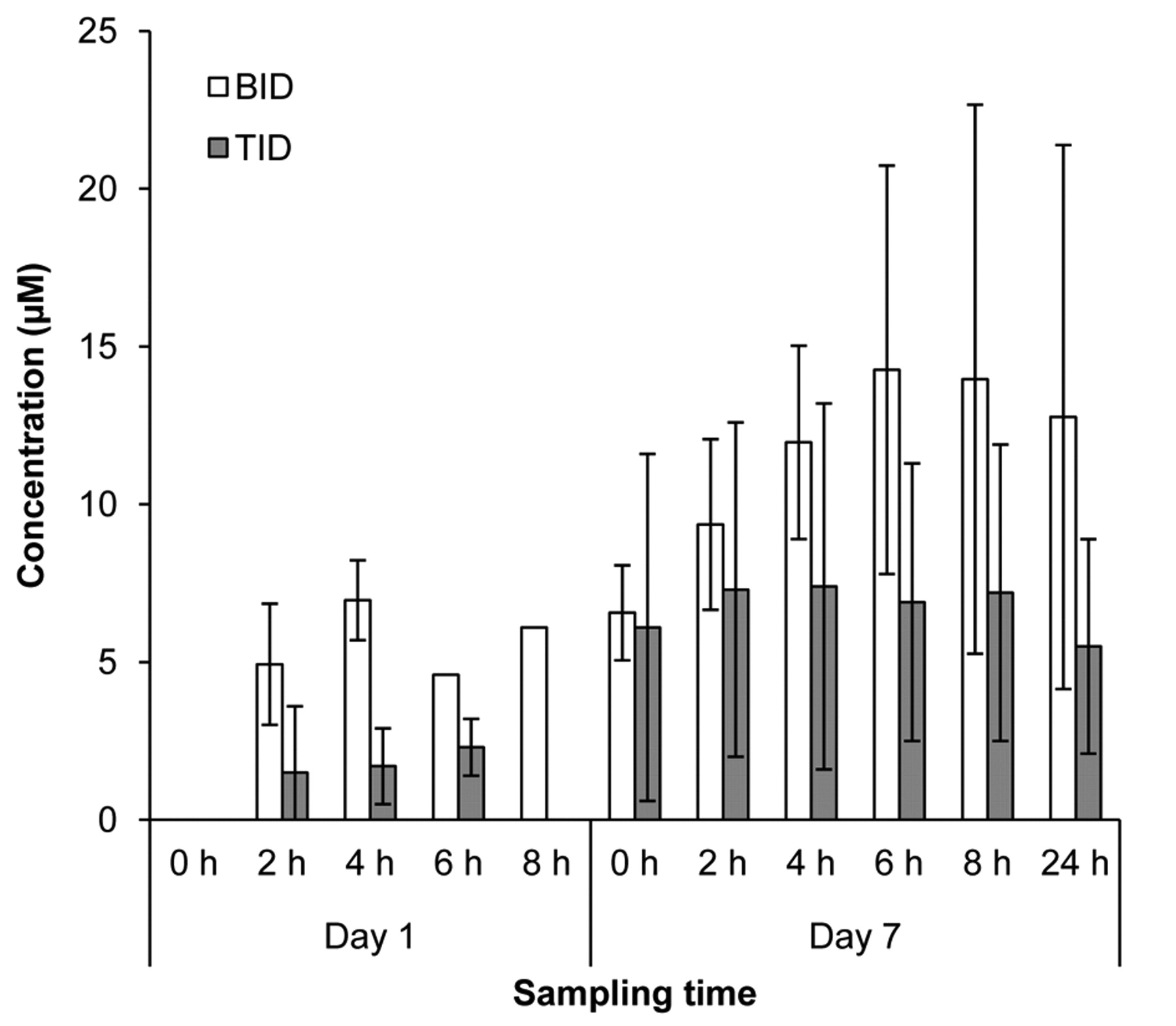
Pharmacokinetics. Variability in plasma exposures was observed across all dose levels evaluated, with some patients achieving exposures above the targeted 9 μM level associated with both activity in preclinical models and efficacy in relapsed pediatric neuroblastoma using the same fenretinide/LXS oral powder formulation (14). 4-HPR was detectable in the first sample collected after the morning dose (2 h) for all patients from whom samples were collected (data not shown). Because of DLTs and patient withdrawals, pharmacokinetic data could only be collected over cycles 1 and 2 from the first three patients enrolled into Dose Level 1 (Figure 1A). Peak exposure levels for all three patients receiving 1,000 mg/m2/day administered in two divided doses exceeded 10 μM, and exceeded 20 μM in one patient who received 24 cycles of therapy. Interestingly, mean peak plasma levels appeared lower in patients receiving 1,300 mg/m2/day, possibly due to decreased absorption from intestinal irritability (Figure 1B). Mean and peak 4-HPR levels were lower in patients receiving 1,000 mg/m2/day divided into three rather than two doses over 24 h (Figure 2). Similar exposures were measured in patients on the MTD of 800 mg/m2/day divided into three daily doses (Figure 3).

4-HPR and 4-MPR plasma levels in cycle 1 for: A) patients 1-3 (Dose Level 1, 1,000 mg/m2/day, divided into two doses); and B) patients 4-6 (Dose Level 2, 1,300 mg/m2/day, divided into two doses). Sampling times are in reference to the morning dose.
Adverse events. CTC grade ≥2 felt to be related to study medication.
Discussion
Fenretinide has demonstrated promising activity in preclinical models of various tumor types. In earlier evaluations, fenretinide was formulated in a corn oil-containing capsule, resulting in variable and low systemic exposures. The formulation evaluated in the current clinical trial was designed to improve systemic exposure, reduce interpatient variability, and potentially improve patient compliance compared with the previous corn oil formulation. Mean plasma levels of fenretinide on the doses and schedules evaluated in our trial were generally no better than those achieved with higher doses (1,800 mg/m2/day) of the oral corn oil-containing capsule evaluated in adults with refractory solid tumors, although compliance was also an issue with the large capsules used in those studies (7, 9). Two trials have been conducted with the current fenretinide/LXS formulation, one in pediatric patients with relapsed neuroblastoma (14), and the present study in adult patients with refractory solid tumors. In the pediatric population, fenretinide/LXS oral powder administration resulted in higher mean peak plasma exposures and was well tolerated. However, in the present adult population, the study drug was less well tolerated, with GI complaints limiting dosing. In spite of the dosing regimen being modified to administer the agent in three divided doses instead of two, the regimen still caused toxicities that resulted in the dose being de-escalated to 800 mg/m2/day in three divided doses, which was established as the adult MTD for this schedule. Adult patients appeared to be particularly sensitive to the GI side-effects of the LXS matrix at higher doses. These side-effects were formally mild by Common Toxicity Criteria grading, but resulted in 7 out of the 20 patients withdrawing from the study.

4-HPR plasma levels in cycle 1 for patients administered 1,000 mg/m2/day divided in two doses (BID, patients 7-9) or three doses (TID, patients10-13). Sampling times are in reference to the morning dose.
One of the most common clinical toxicities of fenretinide has been nyctalopia associated with plasma retinol levels below 100 ng/ml (6). Rodent and human clinical data suggest that fenretinide can displace retinol from circulating serum retinol-binding protein (RBP), leading to a deficiency state in the retina (15). Fenretinide also decreases serum retinol levels by decreasing serum RBP and RBP–retinol complex release from the liver (16, 17), and may increase RBP sequestration in the kidney (18), resulting in nyctalopia. This side-effect is transient, reversible (upon drug cessation or a ‘drug holiday’), and has not caused significant noncompliance. In the present study, 4 out of our 20 patients developed nyctalopia (3 patients had grade 1, 1 patient had grade 2), with 3 of these patients developing symptoms starting in cycle 1, as expected from prior experience with the study drug. The total duration of nyctalopia developing during the 7-day treatment period ranged from 5 to 10 days and resolved within a few days of stopping drug administration in each cycle. One patient received a total of 24 cycles and experienced transient worsening of nyctalopia from grade 1 to 2 during cycle 6 with symptoms lasting for 10 days until complete resolution. In all subsequent cycles, this patient experienced fewer than 10 days of grade 1 nyctalopia per cycle. No patients withdrew from the study due to nyctalopia.
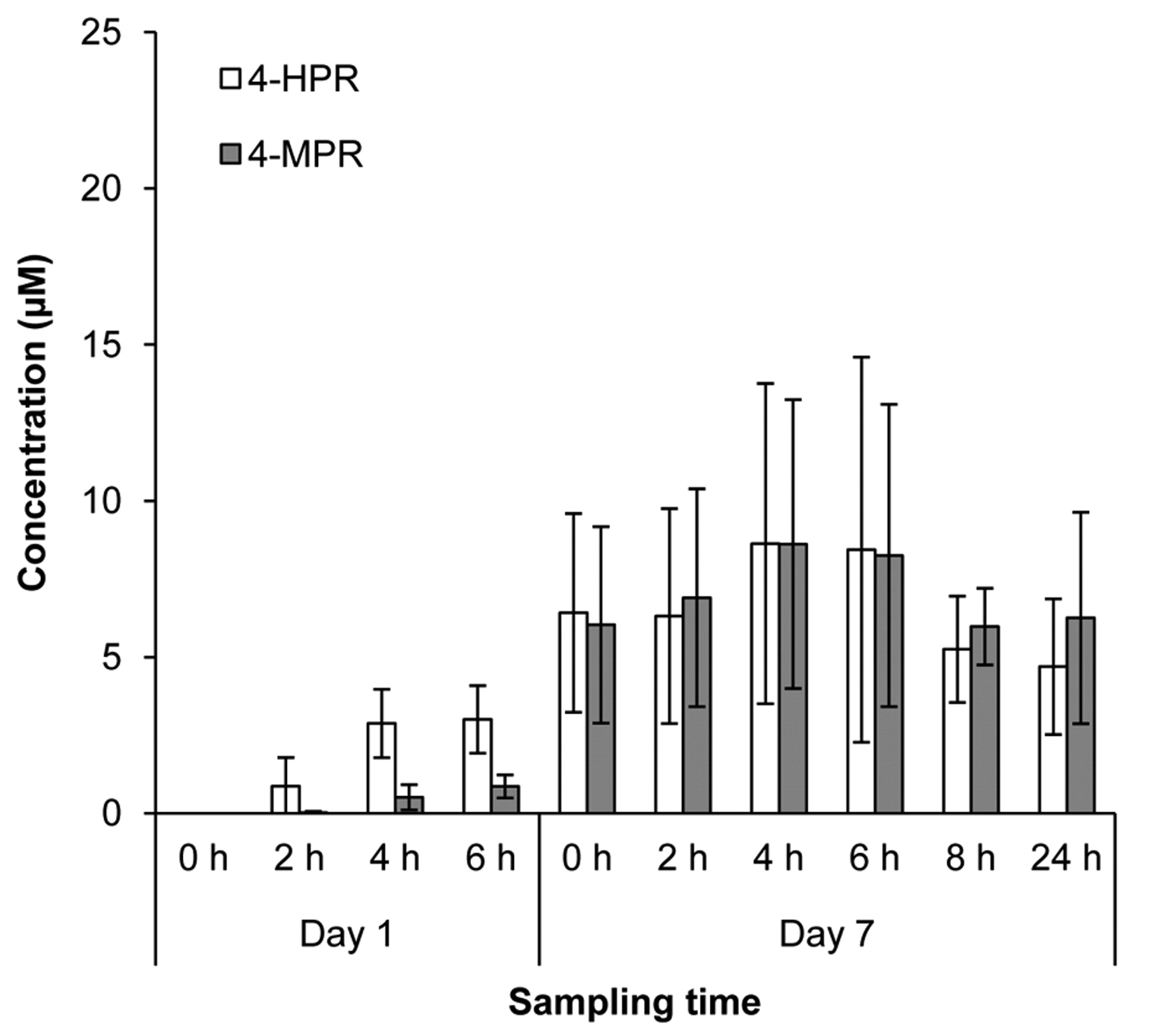
4-HPR and 4-MPR plasma levels in cycle 1 for patients 14-20 (Dose Level −1, 800 mg/m2/day, divided in three doses). Sampling times are in reference to the morning dose.
The present study is the first reported clinical experience utilizing large volumes of LXS in adult populations. GI irritation limited the consistent delivery of higher plasma levels of fenretinide to adult cancer patients using the present fenretinide/LXS oral powder. Discussions with the manufacturer regarding modification of the LXS composition to minimize the observed GI irritation are ongoing.
Acknowledgements
This research was supported by the Center for Cancer Research and the Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institutes of Health; by a Developmental Therapeutics Program, Rapid Access to Intervention Development award to BJM; and by Grant RP10072 from the Cancer Prevention and Research Institute of Texas to CPR. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
We thank Dr. David W. Yesair, BioMolecular Products, Newburyport, MA, USA for advice with the LXS formulation. Lym-X-Sorb matrix was manufactured under license by Avanti Polar Lipids, Alabaster, AL, USA. Certain intellectual property rights to fenretinide/LXS oral powder may be retained by Childrens Hospital Los Angeles, Los Angeles, CA, USA. BJM and CPR may potentially benefit financially from fenretinide/LXS oral powder under the intellectual property policies of Children's Hospital Los Angeles. We thank Gina Uhlenbrauck, SAIC-Frederick, Inc., for editorial assistance in the preparation of this manuscript.
Footnotes
-
This article is freely accessible online.
- Received January 4, 2011.
- Revision received February 18, 2011.
- Accepted February 21, 2011.
- Copyright© 2011 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}