


Abstract
Background: The endothelin axis has recently emerged as an important factor in tumour metastasis. The aim of this study was to investigate the endothelin axis and its downstream pathways related to metastasis in colon carcinoma. Materials and Methods: mRNA expression of 36 genes associated with the endothelin axis in 18 non-metastatic and 20 metastatic colon carcinomas with individual-matched normal mucosa were evaluated using real-time reverse transcription polymerase chain reaction. Results: Seventeen out of 36 genes, including endothelin A receptor, were significantly overexpressed in the tumour tissue compared to the individual-matched normal mucosa. Seven out of 36 genes, including endothelin B receptor, were significantly down-regulated in tumour tissue. Phosphatase and tensin homolog (PTEN) was significantly down-regulated in the metastatic patients compared to the non-metastatic patients. Conclusion: This study indicated that central genes in the endothelin axis are overexpressed when colon tissue becomes malignant. Down-regulation of PTEN may promote a progressive phenotype of colon carcinomas.
- Colon cancer metastasis
- epithelial-mesenchymal transition (EMT)
- endothelin axis
- endothelin A and B receptor (ETAR and ETBR)
- phosphatase and tensin homolog (PTEN)
The majority of colorectal tumours develop along the adenoma-carcinoma sequence which includes gene mutations or epigenetic changes, resulting in alterations of oncogenes, tumour suppressor genes and mismatch repair genes (1, 2). Despite a high five-year survival rate in early colorectal cancer, treatment of patients with distant metastases is still associated with low survival rates (3). Development of new treatment protocols depends on improved knowledge of the molecular mechanisms controlling tumour progression, especially those processes that facilitate the switch from a localised neoplasia to an invasive and metastatic disease. The initial stage of the metastatic process of cancer is associated with morphogenetic changes referred to as the epithelial–mesenchymal transition (EMT) (4). During EMT, cells lose many of their epithelial characteristics, such as cell cell adhesion, polarity and lack of motility, and take on properties that are more typical of mesenchymal cells. EMT enables tumour cells to migrate and to form metastases. Through multiple different signalling pathways, EMT is triggered by a diverse set of stimuli including hypoxia, tumour–stromal cell interactions and growth factor signalling. Targeting specific growth factors or certain critical signal transduction pathways involved in the metastatic cascade has recently contributed to the growing development of novel therapies for colorectal cancer patients (5, 6).
Endothelins (ETs) have recently emerged as important factors in tumour growth and metastasis by regulating EMT, in addition to regulating angiogenesis, cell survival, invasion and metastatic dissemination in several tumour types (7-12). Endothelin-1 (ET-1) is a powerful mitogenic and anti-apoptotic peptide produced by many cancer cells. It exerts its effects by binding to two distinct cell-surface ET receptors, ETAR and ETBR, referred to as the ET axis (13). ET-1 acts as an autocrine growth factor selectively through the ETAR triggering the attendant activation of multiple pathways. The activation of the ETAR pathway by ET-1 may indirectly lead to the stimulation of the AKT survival pathway and mammalian target of rapamycin (mTOR) signalling pathway, and the parallel stabilisation of the zink finger protein snail and β-catenin that translocate to the nucleus to engage transcriptional programs leading to EMT. In ovarian cancer the ET axis has been shown to drive EMT by inducing an invasive phenotype through the down-regulation of epithelial markers such as E-cadherin, in addition to increased levels of β-catenin, snail, N-cadherin and other mesenchymal markers (14, 15).
The complex ET axis has not previously been evaluated in colon cancer patients. However, a significantly elevated ET-1 expression has been shown in 80% of primary human colon carcinomas (16, 17). Furthermore, several studies have shown that both ET-1 and ETAR tend to have increased expression in colon adenomas and adenocarcinomas compared with healthy colon (18-20). In rat colon cancer cells, ET receptor blockade has shown to potentiate FasL-induced apoptosis, while in vivo, however, mixed ETA/ETB receptor antagonism did not control tumour progression (19). Nevertheless, in another rat model a reduction of colorectal cancer liver metastases was seen by ETAR antagonism, indicating ETA antagonists as potential anticancer agents (17). Specific ETAR antagonists have been included in clinical trials in several types of cancer as a potential treatment strategy, but still not in colon cancer patients (21-23).
The PI3K signalling pathway is an important driver of cell proliferation and cell survival and it cross-talks with essential elements in the ET axis, since the activation of ETAR by ET-1 is dependent on PI3K. The PI3K signalling pathway has been shown to contribute to tumour initiation and progression in many types of human malignancies (24). The tumour-suppressor phosphatase with tensin homology (PTEN) negatively regulates the PI3K signalling. This is important since it results in inhibition of downstream signalling pathways such as the AKT pathway, the mTOR signalling pathway and the ET axis, each of which triggers cell survival, cell growth, angiogenesis and invasion through EMT. Recently, PTEN loss has been shown to induce EMT in human colon cancer cells (25). Moreover, alterations in the PI3K pathway have been identified in colorectal cancer (24, 26) and individual components of this pathway have been suggested both as prognostic factors as well as therapeutic targets for drug development (27, 28). The knowledge of the ET axis and associated pathways in colon cancer is important for understanding the key mechanisms of the complex signalling network that programme metastasis establishment and secondary tumour formation in colon cancer patients and possibly for the detection of new therapeutic targets. Expression of genes in the ET axis and its associated pathways in metastatic and non-metastatic colon cancer patients using real-time reverse transcription polymerase chain reaction (qRT-PCR) has not been analysed to date.
The current study examined significant differences in the expression of 36 genes associated with the ET axis and its downstream interactions, within a series of non-metastatic (Dukes B) and metastatic colon carcinomas (Dukes C with metastasis within 5 years or Dukes D), including individual-matched normal mucosa. Additionally, significant differentially expressed genes were further evaluated at protein level to determine the concordance with mRNA and the distribution of ET-1-initiated signalling pathways in the metastatic process of colon cancer.
Materials and Methods
Patients and tissue specimens. After receiving informed consent, tumour tissue samples and individual-matched normal mucosa were obtained from 38 patients with colonic adenocarcinoma who underwent resection at Akershus University Hospital Trust (Lørenskog, Norway) between 2004 and 2009. The dissected tissue samples were collected in the operating room and stored immediately in approximately 5 volumes of RNAlater (Ambion Inc., Austin, TX, USA) and frozen at −80°C. Eighteen patients with non-metastatic disease, Dukes B (with a minimum of 12 negative lymph nodes) and 20 patients with distant metastasis during five-year follow-up (Dukes C) or patients classified as Dukes D were included in the study. There were 22 women and 16 men, with a mean (±standard deviation) age of 69±14 years (range, 29-92 years) at surgery. Three sectioned pieces of the tumour samples were made. The central piece was further processed for RNA isolation, while the two end pieces were fixed in formalin and embedded in paraffin (FFPE). Four-μm sections of FFPE samples were stained with Hagen's haematoxylin and examined by a pathologist for determination of percentage tumour cells. Tissue samples with more than 70% tumour cells were considered as tumour samples and included in the study.
mRNA isolation. Total RNA isolation was performed using the method of Wei and Khan (29) modified to also include miRNA for further analyses. Approximately 60 mg frozen tissue was homogenised in TriReagent (Ambion Inc.) using Mixer Mill MM301 (Retsch, Haan, Germany) for 2×2 min at 30 Hz. After phase separation with chloroform, the aqueous phase (containing RNA) was mixed with 1.5 volumes 100% ethanol and transferred to an RNeasy Mini spin column (Qiagen, Düsseldorf, Germany). Further processing was performed following the manufacturer's protocol. A DNase treatment was included in the procedure. RNA was eluted in 60 μl RNase-free water and stored at −80°C. The concentration of each RNA sample was obtained from A260 measurements using the NanoDrop 2000 (Thermo Fischer Scientific Inc., Waltham, MA, USA). The RNA integrity number (RIN) was tested using the Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA, USA).
Complementary DNA (cDNA) synthesis. cDNAs were produced from 1 μg RNA of each sample using the High Capacity RNA-to-cDNA Master Mix (Life Technologies Corp., Carlsbad, CA, USA) according to the manufacturer's instructions. The following thermal cycler conditions were used: 5 min at 25°C, 30 min at 42°C and 5 min at 85°C. Three random RNA samples were additionally run in the absence of reverse transcriptase enzyme to assess the degree of contaminating genomic DNA. Real-time PCR with genomic DNA-specific assay revealed that RNA was free of genomic DNA (data not shown).
TLDA design and preparation. In order to identify suitable reference genes in the examined tissue, Human Endogenous Control Arrays (Life Technologies Corp.) were used. TaqMan Endogenous Control Arrays are 384-well microfluidic cards containing 16 gene-expression assays commonly used as endogenous controls or genes that exhibit minimal differential expression across different tissues. cDNA from tumour and healthy tissue samples from eight randomly chosen Dukes B patients and eight randomly chosen Dukes C/D patients were included on the arrays. According to geNorm, IPO8, B2M and 18S were the most suitable reference genes in the selected cohort of tissue samples and these were further included at the custom designed TaqMan Low Density Array (TLDA) (Life Technologies Corp.). The TLDA consists of eight separate loading ports that feed into 48 separate reaction chambers for a total of 384 wells per card. Each chamber well contains specific, user-defined primers and probes that are capable of detecting a single gene. The TLDA card that was used in this study was configured into eight identical 48-gene sets. Genes were chosen due to their association with the ET axis and the EMT in colon cancer (Table I). A quantity of 50 μl cDNA (1 μg mRNA) was used as a template. Matched samples from four patients were loaded on each card. No template control (NTC) was added in one loading port. PCR amplification was performed using the ABI Prism 7900HT Real Time PCR System (Perkin-Elmer Applied Biosystems, Foster City, CA, USA). Thermal cycling conditions were used as follows: 2 min at 50°C, 10 min at 94.5°C, 30 s at 97°C and 1 min at 59.7° C for 40 cycles.
ET axis gene expression in tumour tissue versus healthy tissue in colon cancer patients.
Western blotting. Proteins were isolated from the remaining organic phase after isolation of nucleic acids by precipitation with ice-cold acetone for 10 min at room temperature (RT). The pellet was washed three times with 0.3 M guanidine hydrochloride in EtOH at RT. The last wash was performed in 2.5% glycelrol in EtOH for 10 min at RT. The pellet was air-dried for 10 min at RT and dissolved in lysis buffer (25 mM Tris, pH 7.5, 1% NP-40, 100 mM NaCl, 20 mM NaF, 1 mM orthovanadate 1 μg/ml leupeptin, 1 μg/ml antipain, 1 μg/ml pepstatin A and 1 μg/ml chymostatin) and an equal volume of 2× SDS sample buffer was added. The samples were subsequently boiled for 5 min. Following resolution on SDS-polyacrylamide gels, polypeptides were transferred to PVDF membranes by electroblotting. The membranes were blocked for 1 h with 5% non-fat dry milk and then probed with respective antibodies anti-PTEN (A2B1), anti-ETAR (H-60), anti-ETBR (H-74), and Ribosomal Protein LP0 (RPLP0) (T-18), all from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The primary antibodies were detected with HRPO-conjugated secondary antibodies (Southern Biotechnology Associates, Inc., Birmingham, AL, USA). Super-Signal (Pierce Biotechnology, Inc., Rockford, IL, USA) was used as substrate for HRPO and chemiluminescence were detected by a CCD camera (Kodak Image Station 4000MM PRO; Carestream Health, Inc., Rochester, NY, USA). The software CareStream (Carestream Health, Inc.) was used to identify and estimate the intensity of bands. The expression level of the target protein was related to the protein level by calculating the ratio between the band intensity of the target protein and that of the control protein RPLP0.

Significantly overexpressed genes in tumour versus healthy tissue in colon cancer patients.
TLDA and statistical analyses. RealTime Statminer 3.0 Software (Integromics, Madrid, Spain) was used for implementation of quality controls in addition to calculation of differentially expressed genes. This programme uses the comparative Ct method for relative quantification analysis and the results are expressed as fold change of expression levels (ΔΔCt values). Medians were used to replace missing values that occurred due to inconsistencies between replicates rather than from low expression. The detectability threshold was set to 36, meaning that failing detectors were those with Ct ≥36. According to geNorm, IPO8 and 18S were identified as the most suitable pair of reference genes when all the TLDAs were analysed. Significant differentially expressed genes were calculated by parametric tests and the Benjamini-Hochberg procedure was used to control the false discovery rate (FDR). This method assumes that detectors are expressed in a correlated mode. The adjusted p-values are displayed in Table I. To evaluate the Western blot analyses, non-parametric tests were performed. The Wilcoxon test was used to examine the association between ETAR/ETBR protein expression and healthy/tumour tissue. The relationships between PTEN protein expression and non-metastatic and metastatic disease were evaluated using the Mann-Whitney test. A difference of p<0.05 was considered statistically significant. Except for the analyses in RealTime Statminer, all other calculations were performed using SPSS (version 18.0; SPSS, Chicago, IL, USA).
Research ethics. This project was approved by the Regional Committee for Medical Research Ethics, Eastern Norway. The Norwegian Social Science Data Service has approved the data collection and analysis.
Results
Table I shows the data of relative gene expression (RQ) from tumour and healthy tissue in the total group of colon cancer patients, indicating that 17 out of 36 genes associated with the ET axis were significantly overexpressed in the tumour tissue compared to the individual-matched healthy tissue. Among these, GSC, TWIST, LEF1, SNAIL, Cyclin D1, EDNRA, FOXC1 and TCF3 were expressed >two-fold with respect to the healthy tissue, as shown in Figure 1. Only 7 out of 36 genes were significantly down-regulated in tumour tissue compared to the healthy tissue. These were EGF, EDNRB, ZEB2, PTEN, PIK3CD, PIK3R1 and CDH1, as illustrated in Figure 2. When the tumour tissues in the non-metastatic and in the metastatic cancer patients were compared, only one gene was significantly differentially expressed. As shown in Figure 3, PTEN was 0.53-fold down-regulated in tumour tissue from patients with metastatic compared to non-metastatic colon cancer. A summary of the differentially expressed genes in the ET axis and its associated pathways is illustrated in Figure 4.

Significantly down-regulated genes in tumour versus healthy tissue in colon cancer patients.
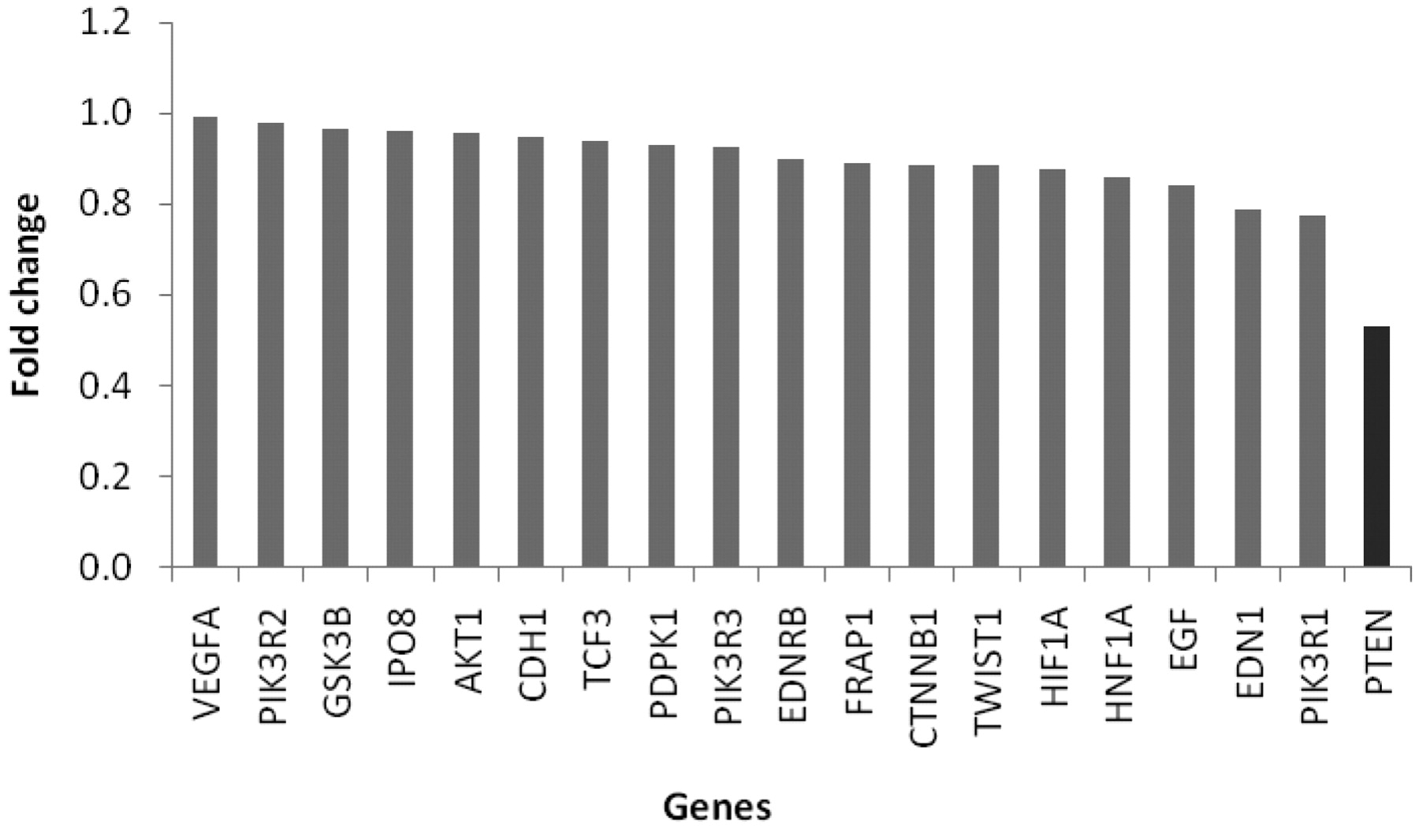
Down-regulated genes in tumour tissue in metastatic versus non-metastatic colon cancer patients. PTEN is significantly differentially expressed (p=0.0172).
A total of 16 tumour tissue samples, eight from Dukes B and Dukes C/D colon cancer patients respectively were randomly selected for protein detection of PTEN. The protein level of PTEN using Western blot analysis revealed no significant down-regulation in the patients with metastatic disease compared to non-metastatic disease (0.613) (Table II). The gene expression data showed that the gene for one of the ET receptors, ETAR, was overexpressed in the tumour tissue, while that for the other ET receptor, ETBR, was overexpressed in the healthy tissue. In order to examine the expression pattern of the two ET receptors, their corresponding proteins were evaluated in tumour and healthy tissue in eight randomly selected colon cancer patients (Table III). Neither ETAR nor ETBR protein showed significant differences between tumour and healthy tissue (0.401 and 0.123).
Discussion
Different signalling pathways have been shown to trigger EMT and the search for the molecular mechanisms controlling it is ongoing. The present study revealed that 17 out of 36 examined genes in the ET axis and its downstream pathways were significantly overexpressed in tumour tissue compared to individual-matched healthy mucosa in colon cancer patients. Furthermore, seven genes were down-regulated in the tumour tissue compared to the healthy mucosa. A shift of gene expression from ETBR to ETAR was found when colon tissue became malignant. When comparing tumour tissue in the non-metastatic and metastatic colon cancer patients, one gene, PTEN, was significantly differentially expressed, showing a down-regulation in the metastatic cancer patients.
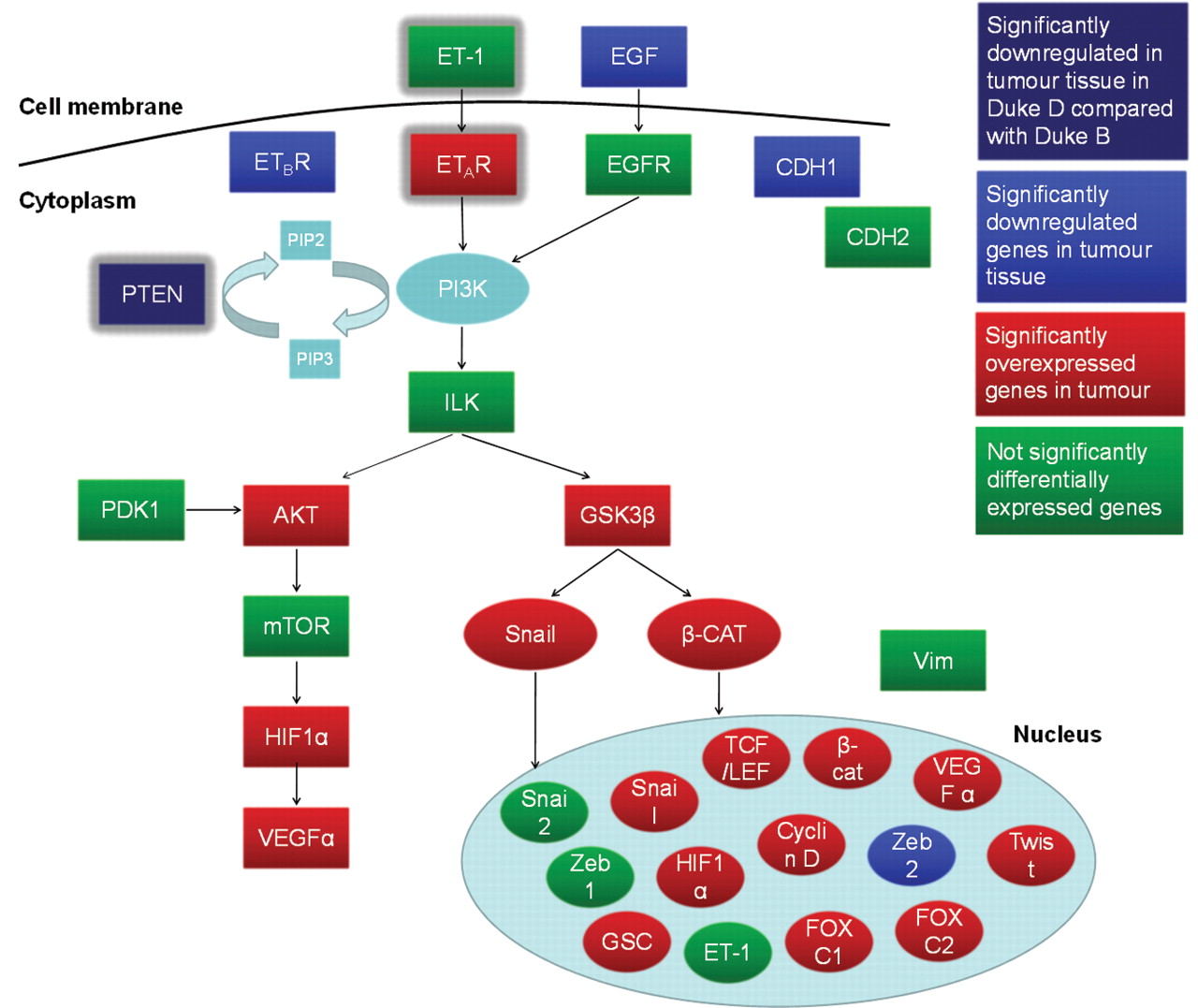
The ET axis and its associated pathways. The figure shows differentially expressed genes between tumour and healthy tissue in the total patient group (blue and red) and differentially expressed gene (PTEN) between tumour tissue in metastatic and non-metastatic colon cancer patients (indigo blue).
The TLDA analyses of tissue samples in this study provided a more standardised experimental setup compared to established single qRT-PCR assays. TLDA analyses require relatively small amounts of cDNA and PCR reagents and are less time- and labour-intensive than routine qRT-PCR (30). Using this setup and dedicated software for analysing data (RealTime Statminer 3.0 Software), this approach allowed the efficient and standardised detection of expression of large number of genes in a large number of samples at the same time (31).
This is the first study to show a shift in expression of the ET receptors at the gene level in colon carcinomas corresponding to the malignant potential in the tumour. The gene for ETAR was overexpressed in the tumour tissue compared to the healthy tissue, while that for the other subtype of the ET receptors, ETBR was down-regulated. One possible mechanism for the ETBR down-regulation at the mRNA level may be by methylation of the EDNRB gene, which has previously been reported in colorectal cancer specimens (32). The protein verification of the ET receptor, however, in the present study did not reveal any significant differences. The results of this study are in contrast to the findings of Egidy et al., who demonstrated increased protein and mRNA levels for both receptor subtypes in colorectal cancer compared with healthy tissue (20). Hoosein et al., however, observed a shift in ET receptors at the protein level using autoradiography (33). They demonstrated that ETAR binding was up-regulated, whereas ETBR binding was down-regulated in primary colorectal cancer compared with healthy colon. The observed shift from ETBR to ETAR in this study indicates an up-regulation of the ET axis and its downstream pathways when colon tissue becomes malignant. However, the gene for growth factor ET-1, which binds to the ET receptors and activates downstream pathways, was not found to be significantly differentially expressed. Nevertheless, the ET axis may be overexpressed in the tumour tissue due to the shift in ET receptors rather than because of increased ET-1 expression when colon tissue becomes malignant. An activated ET axis in the tumour tissue is supported by the gene alterations in its downstream pathways identified in the current study.
Intensity ratio from Western blot analyses between PTEN and the control protein RPLP0 in tumour tissue from patiens with non-metastatic and metastatic colon cancer. Comparison of the ratios between the two groups yielded a p-value of 0.613 (Mann-Whitney test).
In the AKT pathway, AKT, HIF1A and VEGF showed an increased mRNA expression in the tumour tissue compared to the healthy tissue, indicating increased angiogenesis and cell survival. HIF1α normally activates the transcription of genes encoding VEGF, ET-1 and transferrin, which are implicated in vasodilation, neovascularisation and tumour metastasis (34, 35). In the present study, increased levels of HIF1A may have influenced the increased level of VEGF. However, the increased levels of HIF1A seem not to have influenced the levels of ET-1 in these patients, since no significant difference of ET-1 was observed between tumour and healthy tissue. Increased expression of genes in the AKT pathway have previously been demonstrated in colorectal cancer, however, not in the context of the shift in the ET receptors, as shown in the present study (24).
In addition to AKT, the activated ET axis observed in the tumour tissue may affect other downstream protein kinases, for example, causing inhibition of GSK-3β. GSK-3β has the ability to phosphorylate snail, β-catenin and cyclin D1, resulting in the destruction of these factors. When GSK-3β is inhibited, the level of snail, β-catenin and cyclin D1 is enhanced. The present results showed a slight increase of GSK-3β (GSK3B) when comparing tumour and healthy tissue which may not indicate an inhibition. Despite this increase, the expression of SNAI1, CTNNB1 β-catenin and cyclin D1 (CCND1) is comparatively higher than that of GSK3B. As a consequence, their increase helps to induce the expression of a large constituency of genes, among them those specifying several EMT-inducing transcription factors (36). The overexpression of the transcription factors GSC, TWIST, LEF1, TCF3, FOXC1 and FOXC2 observed in this study has been shown to contribute to the establishment and maintenance of mesenchymal cell phenotypes, which again facilitates migration and formation of metastases (37, 38).
Intensity ratio from Western blot analyses between the control protein RPLP0 and ETAR and ETBR respectively, in healthy and tumour tissue. Comparison of the ratios between the two groups yielded p-values of 0.401 and 0.123, for ETAR and ETBR, respectively (Wilcoxon test).
In the present study, E-cadherin (CDH1) was found to be significantly down-regulated in tumour tissue compared to healthy mucosa. The increase of different transcription factors observed in the tumour tissue may have an impact on the transcription of other genes identified in this study, including CDH1. SNAI1, SNAI2 and TCF3 are known to function as direct transcriptional repressors of E-cadherin expression, while others, such as TWIST, ZEB1, FOXC1, FOXC2 and GSC act less directly on E-cadherin (37, 38). In the present study the majority of them (SNAI1, TCF3, TWIST, FOXC1, FOXC2 and GSC) were overexpressed in the tumour tissue compared to the healthy tissue, hence they may be responsible for the E-cadherin down-regulation. E-Cadherin down-regulation has previously been reported in colon cancer tissue and some studies have even demonstrated that reduced E-cadherin expression in the primary site is directly proportional to the frequency of lymph node and liver metastasis (39, 40). In contrast to the present results, Truant et al. did not find any statistically different level of E-cadherin when comparing colon carcinomas with adjacent healthy mucosa (41). Conversely, they found that E-cadherin levels in liver metastases are significantly higher than in adjacent healthy tissue, indicating that E-cadherin expression changes dynamically in colon carcinomas.
In addition to E-cadherin, PTEN was also significantly down-regulated in the tumour tissue compared to the healthy mucosa. In healthy tissue, PTEN acts as a tumour suppressor gene and suppresses the PI3K/AKT pathway, hence it also inhibits the ET axis (42). The present results showed that when colon tissue becomes malignant, the gene expression of PTEN is decreased. As a consequence, the downstream pathways of the ET receptors are more active and this may increase angiogenesis, cell survival, cell proliferation, EMT and invasion, all of which facilitate the progression of colon carcinomas. Interestingly, in the present study PTEN was the only gene out of the 36 examined genes that showed significant differential expression when comparing the tumour tissue in patients with metastatic colon cancer that with in non-metastatic disease. This is the first study to show that PTEN is significantly down-regulated in patients with metastatic disease (Dukes C with metastasis within five years and Dukes D) compared to non-metastatic disease (Dukes B). Jiang et al. reported similar results in a study showing that the mRNA level of PTEN expression was significantly higher in Dukes A or B than in Dukes C or D (43). Protein verification of PTEN, however, has been evaluated in several studies during the last decade. In the present study, protein verification of PTEN did not reveal any significant difference between the tumor tissues in the two groups of patients. In contrast to these results, loss of PTEN protein or function has been shown in a large fraction of advanced colorectal cancers, indicating that uncontrolled signalling through PI3K contributes to metastatic cancer (42, 44-46). Taken together, these results indicate that down-regulation of PTEN is involved in the pathogenesis, invasion and metastasis of colon carcinomas. Lower or loss of PTEN expression has also been shown to reduce the survival rate, indicating PTEN as a possible prognostic marker in colon cancer (44, 46). Additionally, PTEN expression has recently been suggested as an indicator of therapy resistance in this type of cancer (28, 47-51).
In the present study, the down-regulation of PTEN and the shift in the ET receptors in colon carcinomas indicated an up-regulation of the ET axis and its associated pathways. Both changes facilitate cell proliferation, cell survival and EMT in colon cancer tissue. PTEN may be a key hub among several major pathways which regulate the progression of colon carcinoma. Additionally, the study demonstrated that several key transcriptional factors are regulated in the tumour tissue, facilitating EMT and the formation of metastases. The gene alterations identified in this study may add to the understanding of the metastatic process of colon cancer and some of them, such as the ET receptors and PTEN, may be used as biomarkers for predicting colon cancer progression as well as potentially therapeutic targets, although they still need further validation in prospective diagnostic settings.
Acknowledgements
We thank Department of Clinical Molecular Biology (EpiGen), Akershus University Hospital, Norway, for laboratory facilities, and Randi Bjørseth at Department of Pathology, Akershus University Hospital, Norway, for guidance and technical assistance. This study was supported by grants from South Eastern Norway Regional Health Authority, Norway.
- Received February 6, 2011.
- Revision received February 22, 2011.
- Accepted February 23, 2011.
- Copyright© 2011 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.