


Abstract
Background: “Oncogene addiction” is a concept in which tumor cells exhibit dependence on certain oncogene(s) for their sustained proliferation and survival, thus providing the rationale for molecular targeted therapies. Cancer cells addicted to epidermal growth factor receptor (EGFR) bear activated mutations in the EGFR gene, and these mutations are used as the markers for predicting carcinomas susceptible to EGFR inhibitors such as gefitinib and erlotinib. However, other unknown mechanisms underlying susceptibility to EGFR inhibitors have also been suggested. Materials and Methods: The susceptibility of non-small-cell lung cancer (NSCLC) cell lines to EGFR inhibitors and the pattern of their oncogene addiction was examined. The effect of EGFR inhibitors on the activation of the oncogene was analyzed. The possible use of the oncogene protein expression as a biomarker was assessed. Results: HER2 addicted, non-EGFR expressing NSCLC cell line NCI-H2170 was susceptible to EGFR inhibitors. EGFR inhibitor treatment led to markedly decreased phosphorylation levels of activated HER2 and its downstream effector AKT. Furthermore, the soluble form of HER2 was secreted by NCI-H2170 cells and was positively detected in the blood of xenografted mice. Conclusion: HER2 seems to be a valid therapeutic target of EGFR inhibitors in HER2-addicted lung carcinomas, and soluble HER2 may be an effective biomarker to guide the appropriate treatment of such cancer cells.
Recent progress in molecular cancer therapeutics has led to the development of new antitumor drugs targeting the particular signaling pathways, on which the proliferation and survival of tumors depend. Although the mechanisms of their antitumor effects remain to be elucidated, the concept of “oncogene addiction” is proposed to provide rationale for such molecular targeted therapeutics. When the survival and malignant phenotype of a cancer depend on certain oncogenes, it is regarded as oncogene addicted (1, 2), and agents inhibiting those factors would effectively and specifically damage such carcinomas. The precise diagnosis of oncogene addiction is the key to the success of such therapies.
Epidermal growth factor receptor (EGFR), a member of the transmembrane receptor tyrosine kinase family, is overexpressed in a number of human tumors and its aberrant activation is known to be involved in the development and progression of cancer (3). EGFR is an important target of anticancer agents, and new anti-EGFR inhibitors and monoclonal antibodies are of continued interest in drug development. Small molecular agents such as gefitinib (Iressa™) and erlotinib (Tarceva®) and lapatinib were developed as specific EGFR inhibitors, and now they are used clinically as the antitumor drugs for non-small-cell lung cancer (NSCLCs) (4-6). Throughout the clinical studies, it has been clearly demonstrated that activating mutations in the kinase domain of EGFR are highly correlated with the tumor sensitivity to the agents. Hence, activating mutations such as exon 19 deletions and L858R point mutation have become the most important markers for identifying appropriate patients for such agents. On the other hand, in some cases strong correlation is not present (7-11). To maximize the therapeutic benefits of established EGFR inhibitors in cancer patients, it is essential to clarify the other molecular mechanisms that underlie the susceptibility of carcinomas to the anti-EGFR agents and establish precise predictive diagnostics for their susceptibility to such therapies.
HER2 (also known as ErbB-2, ERBB2) belongs to the same family as EGFR. Its abnormal expression is involved in the progression of some carcinomas including breast cancer (12-14), and HER2 gene amplification is known to enhance the sensitivity of lung carcinomas to gefitinib (15, 16). However, the method by which HER2 confers sensitivity to EGFR inhibitors on tumors remains unclear. According to in vitro kinase assays, gefitinib and erlotinib possess only marginal affinities toward HER2 (17-19), suggesting that the antitumor mechanisms in such malignancies may not be due to direct interactions of EGFR inhibitors with HER2. In contrast to this suggestion, erlotinib effectively inhibited chimeric- and overexpressed-HER2 protein in fibroblast cells with no endogenous EGFR and HER2 expression (20). Since this observation was based on an artificial cell model, further investigations are required to assess whether these EGFR inhibitors possess antitumor potency through direct inhibition of HER2.
In this study, the relationship between HER2 and EGFR inhibitors was examined in NSCLC cells and the possible use of HER2 as a biomarker was investigated.
Materials and Methods
Cells and culture conditions. Human lung carcinoma A549 (ATCC CCL-185), NCI-H460 (ATCC HTB-177), NCI-H1650 (CRL-5883), NCI-H1703 (CRL-5889), NCI-H1975 (CRL-5908), NCI-H1993 (CRL-5909), NCI-H2170 (CRL-5928), HCC4006 (ATCC CRL-2871), and HCC827 (ATCC CRL-2868) were obtained from the American Type Cell Culture Collection. Human lung carcinoma PC-9 was purchased from Immuno-Biological Laboratories Co., Ltd. (Gunma, Japan). The A549 cells were maintained in D-MEM medium (Sigma-Aldrich Corporation, St. Louis, MO, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS). All the other cell lines were maintained in RPMI 1640 medium (Nissui pharmaceutical Co., Ltd., Tokyo, Japan) supplemented with 10% FBS.
Antibodies and reagents. The anti-HER2 and anti-EGFR monoclonal antibodies used for immunoblot, immunoprecipitation and immunohistochemistry analyses were raised against the cytoplasmic domains of human HER2 and EGFR as described previously (21). Anti-phosphotyrosine (PY20) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-AKT mouse monoclonal antibody (2H10) and anti-phospho-AKT S473 rabbit monoclonal antibody (D9E) were purchased from Cell Signaling Inc. (Danvers, MA, USA). Gefitinib, erlitonib and lapatinb were purchased from Tocris Bioscience (Ellisville, MO, USA), LKT Laboratories, Inc., (St. Paul, MN, USA) and Toronto Research Chemicals, Inc., (North York, ON, Canada) respectively, and dissolved in dimethyl-sulfoxide (DMSO) at a final concentration of 10 mM. Small interfering RNAs (siRNAs) targeting human EGFR and HER2 were purchased from Invitrogen (Carlsbad, CA, USA). Stealth RNAi Negative Control (Invitrogen) was used as a control.
MTS cell growth inhibition assay. Growth inhibition by the EGFR inhibitors was assessed by using MTS assay (Promega Corporation, Madison, WI, USA) according to the manufacturer's instruction. The cells were seeded on 96-well plates and after 24 h, the medium was replaced with medium containing 10% FBS and 0.01% DMSO with or without the EGFR inhibitors (gefitinib, erlotinib or lapatinib). Each data point and bar represents the mean value (percentage) relative to untreated cells and standard deviation (n=4), respectively.
Immunoblotting and immunoprecipitation. The cells were harvested with lysis buffer (50 mM sodium phosphate [pH 7.4], 150 mM sodium chloride, 1% NP40 alternative [EMD Chemicals, Inc., San Diego, CA, USA], 1 mM EDTA, 1 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 2 μg/mL leupeptin, 2 μg/mL aprotinin). The lysates were mixed with Laemlli's sample buffer, boiled and 20 μL (40 μg protein) were subjected to SDS-PAGE and blotted onto PVDF membranes. The membranes were blocked, and incubated with primary antibodies (anti-EGFR, anti-HER2 or PY20) and then horseradish peroxydase-conjugated anti-mouse IgG antibody (DAKO, Glostrup, Denmark). The signals were visualized by ECL western blotting detection reagents (GE Healthcare, Buckinghamshire, UK). For immunoprecipitation analysis, 1 mg/mL of the lysate samples were incubated with 2 μg of antibodies and Protein A-Sepharose beads (GE Healthcare) for 60 min at 4°C with agitation. The immunocomplexes were pelleted by centrifugation, washed three times with lysis buffer and resuspended in 40 μL of Laemlli's sample buffer and boiled.
RNA interference. For growth inhibition analysis, each siRNA was transfected using Lipofectamine 2000 (Invitrogen). In brief, 10 pmol of siRNAs dissolved in 25 μL Opti-MEM I (Invitrogen) were combined with 0.25 μL Lipofectamine 2000 reagent, dissolved in 25 μL Opti-MEM I, incubated for 20 min at room temperature and then added to cells in 96 well-plates. After 24 h, the same transfection procedure was repeated. Following another 24 h culture, the medium was replaced. After a total of 96 h incubation, the plates were analyzed for cell proliferation using the MTS assay.
Immunohistochemical analysis. Xenografts were established in nude mice by subcutaneous injection of 106 cells of NCI-H2170 or HCC827. The xenografts whose size exceeded 100 mm3 in 3 weeks were excised, fixed in 10% neutral buffered formalin solution and embedded in paraffin. Then 4 μm thin slices were cut, mounted on slides and incubated with the diluted anti-EGFR or HER2 antibody for 1 h at room temperature. After Tween/PBS wash, the slides were incubated with a horseradish peroxidase conjugated goat anti-mouse antibody (DAKO) for 1 h. Following a Tween/PBS wash, the slides were incubated with liquid DAB substrate-chromogen for 5 min to visualize the presence of antibodies and counterstained with hematoxylin. The slides were observed under a bright-field microscope.
Quantification of soluble HER2 in the serum of tumor-bearing mice by ELISA. Serum samples were obtained from the mice xenografted with NCI-H2170 or HCC827 after they developed tumors >100 mm3. The serum level of HER2 was measured using an ELISA kit (Bender MedSystems, Vienna, Austria) according to the manufacturer's instruction.
Results
Sensitivity to gefitinib and receptor expression. Table I shows the EGFR gene status and sensitivity to gefitinib of the cancer cell lines used in this study (22-24). The sensitivity of these cell lines to gefitinib, as obtained by the present MTS assay was similar to previous reports (Figure 1A): the PC-9, HCC827 and HCC4006 cells were highly sensitive to gefitinib (IC50<0.1 μM), whereas the NCI-H1703 cells were resistant (IC50>10 μM); the NCI-H2170 cells were sensitive to gefitinib (0.5 μM<IC50<5 μM), although the sensitivity of this cell line was lower than PC-9, HCC827 and HCC4006.
EGFR status and gefitinib sensitivity of cell lines used in this study.
Surprisingly, EGFR expression was not detected in the NCI-H2170 cell lysate in contrast to the lysates of the remaining cell lines (Figure 1B). The anti-phosphotyrosine blot displayed no reactive band corresponding to EGFR (approximately 160 kDa) in the NCI-H2170 lysate, but a hyperphospholylated protein was detected around 200 kDa (Figure 1B). To identify this protein, immunoblot analysis was performed based on the assumption that it might be a receptor kinase, based on its low mobility. The anti-HER2 blot showed that the hyperphosphoprotein was HER2 (Figure 1B).
HER2 phosphorylation and specific knockdown effects. To examine the relationship between HER2 overexpression and gefitinib-sensitivity in the NCI-H2170 cell line, the effect of gefitinib on the phosphorylation level of HER2 was examined by immunoprecipitation and immunoblot analyses. As shown in Figure 2, gefitinib abrogated the phosphorylation of HER2 in the NCI-H2170 cells at a concentration of 10 μM. In parallel with the dephosphorylation of HER2, phosphorylation of AKT, one of the key downstream effectors of HER2, was also decreased by gefitinib treatment in the NCI-H2170 cells (Figure 2B). In order to confirm the dependency of NCI-H2170 cell survival on HER2 independently of EGFR, RNAi experiments were performed. Specific knockdown of EGFR and HER2 was observed by immunoblot (Figure 3), and their antiproliferative effects on the cells were assessed by MTS assay. As expected, the proliferation of the EGFR-addicted HCC827 and HCC4006 cells was inhibited by the siRNAs targeting EGFR. The proliferation of the NCI-H2170 cells was inhibited only by the HER2-targeting siRNAs.
Antiproliferative effect of other EGFR inhibitors. Whether erlotinib and lapatinib exert antiproliferative effect on NCI-H2170 cells was examined as well. As shown in Figure 4, although they exhibited different inhibition patterns, both agents inhibited the growth of the NCI-H2170 cells as well as most of the other cell lines.
HER2 secretion. Next, the possibility of using HER2 protein expressed in HER2-addicted cancer cells as a marker to detect cells susceptible to EGFR inhibitors was assessed. Immunohistochemical analysis of the xenografted tumor tissues derived from NCI-H2170 or HCC827 cells showed that the NCI-H2170 tumors exhibited strong HER2 staining, whilst strong EGFR staining was observed in the HCC827 tumors (Figure 5A). In addition, as shown in Figure 5b, an obvious reactive band was observed using anti-HER2 antibody in the lysates and culture supernatants of the NCI-H2170 cells, whereas no detectable band was observed in the HCC827 samples. Furthermore, soluble HER2 was detected only in the serum from mice with HER2-addicted NCI-H2170 cell xenografts (Figure 5C).
Discussion
For EGFR inhibitors such as gefitinib and erlotinib, the activating mutations in the kinase domain of EGFR have been identified and used as the markers (7-10). A part from these mutations, there are no established biomarkers to predict susceptibility to EGFR inhibitors so far.
In squamous NSCLC the activating mutations in EGFR are very rare while erlotinib is often effective on this cancer type (6, 25, 26). By using an artificial non-cancer cell model with no endogenous EGFR and HER2 expression, Schaefer et al. have found that EGFR inhibitor directly inhibited activated HER2 (20). In the present study, a HER2-, not EGFR- addicted NSCLC cell line, NCI-H2170 was susceptible to EGFR inhibitors, probably via the direct inhibition of activated HER2 by the inhibitors. These results suggest that activated HER2 could be used as the marker to predict susceptibility to EGFR inhibitors.
NCI-H2170 is a cell line established from squamous NSCLC which does not bear any mutations in the EGFR gene. Despite this genetic background, this cell line is substantially sensitive to gefitinib. Notably, EGFR expression was not detected in this cell line, indicating that EGFR does not exert oncogenic signaling. Based on the assumption that some other tyrosine kinases might be involved, HER2 was successfully identified as a hyperactivated kinase in this cell line. Since its phosphorylation level was decreased by gefitinib treatment, HER2 seemed to be the direct site of action for gefitinib. The RNAi experiments confirmed that proliferation of the NCI-H2170 cells depended solely on HER2, independently of EGFR, making it a possible target in lung cancer treatment. Thus testing HER2 status in lung cancer patients may help in establishing a more effective therapeutic strategy using EGFR inhibitors such as gefitinib, erlotinib and lapatinib.

Sensitivity of the cell lines to gefitinib and EGFR and HER2 expression status. (A) Mean percentage growth relative to untreated control cells of cells treated with gefitinib for 72 h, assessed by MTS assay. (B) EGFR, HER2 and tyrosine-phosphorylated proteins detected using anti-EGFR, anti-HER2 and anti-phosphotyrosine antibodies, respectively in whole cell lysates.

Effect of gefitinib on receptor phosphorylation. HCC827 control cells (a) and NCI-H2170 cells (b) were treated with gefitinib for 4 h. Whole cell lysates and immunoprecipitants using anti-EGFR mAb (a) and anti-HER2 mAb (b) were subjected to immunoblot analysis with anti-EGFR, anti-HER2, anti-phosphotyrosine, anti-AKT mAbs and anti-phospho-AKT rabbit monoclonal antibodies.
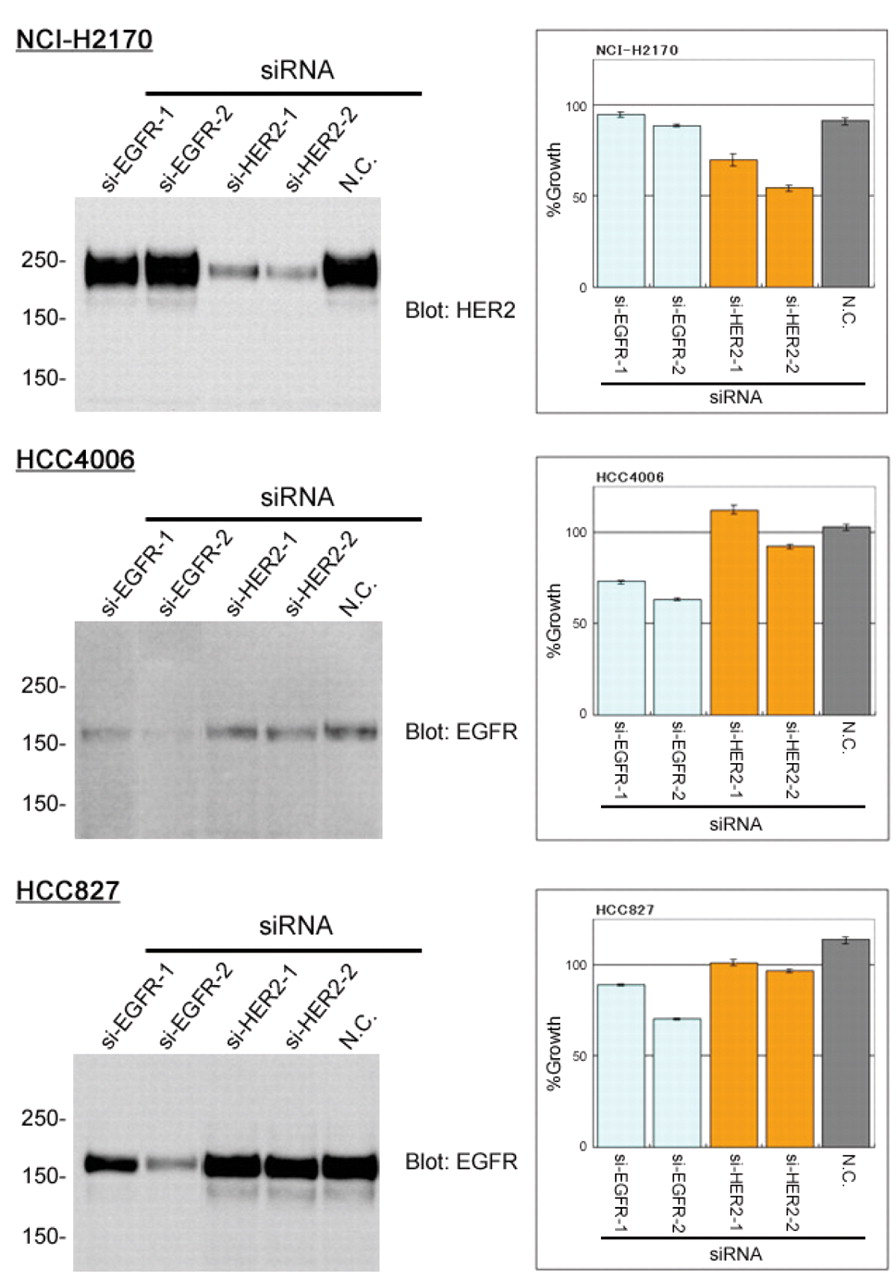
Effects of specific knockdown. NCI-H2170, HCC4006 and HCC827 cells were transfected with siRNA oligonucleotides targeting EGFR or HER2 and the subsequent expression of HER2 or EGFR was analyzed by immunoblotting. Mean cell proliferation relative to mock transfectants (negative control) was assessed with MTS assay.
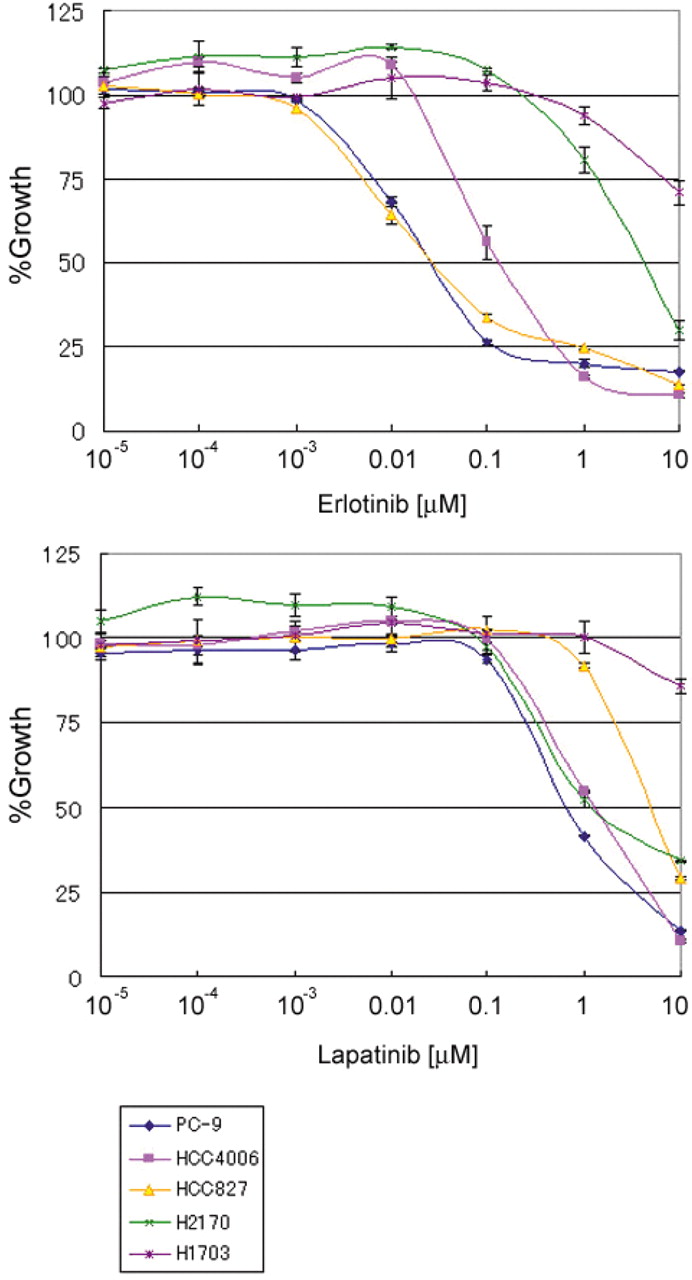
Sensitivity to other EGFR inhibitors. Mean growth inhibition relative to untreated controls of cells treated with erlotinib or lapatinib assessed by MTS assay.
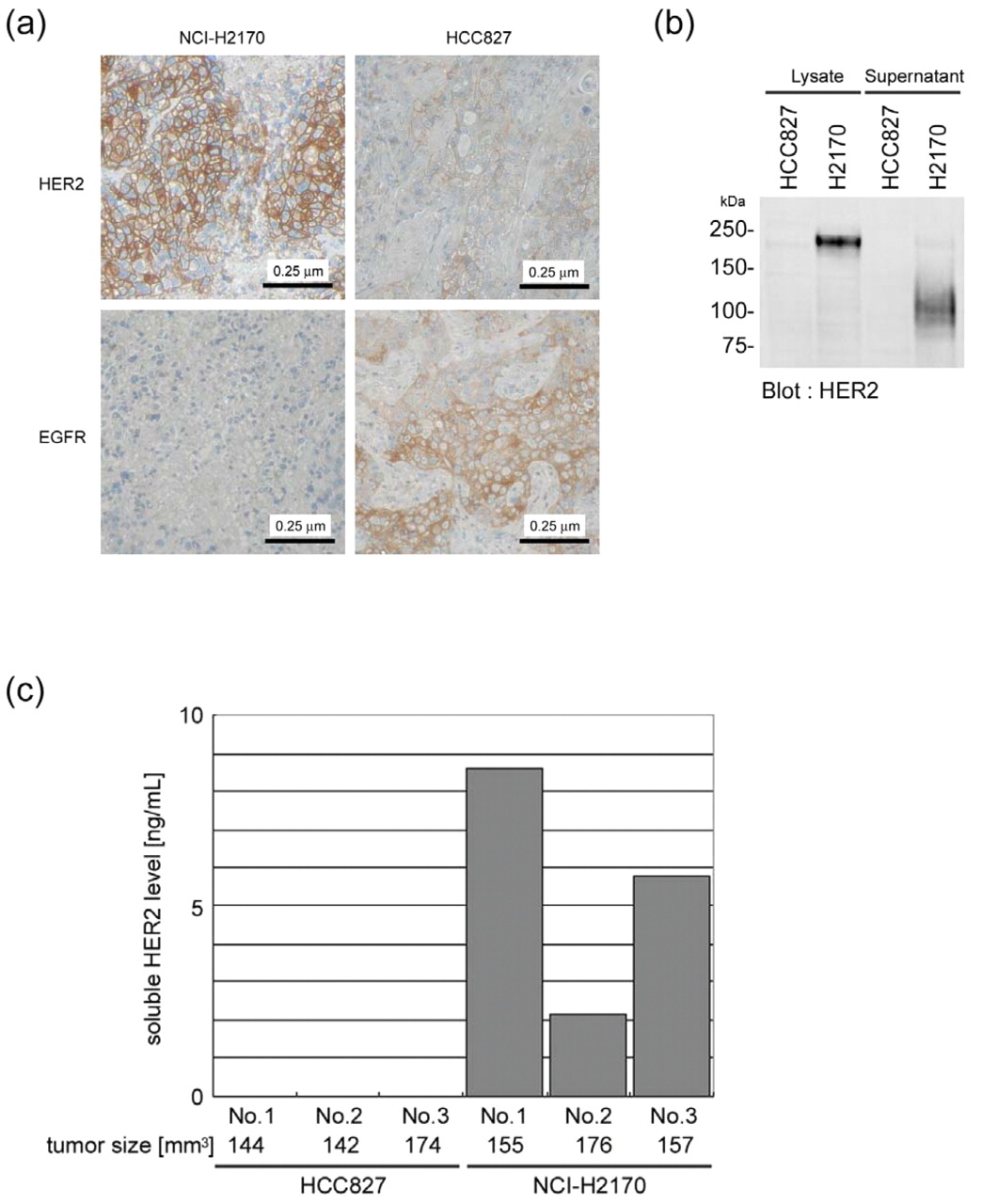
Detection HER2 in vivo. (A) Immunohistochemical analysis of xenografts derived from NCI-H2170 or HCC827 (negative control) cells using anti-EGFR and anti-HER2 antibodies. Nuclei were counterstained with hematoxylin. (B) Immunoblot analaysis of cell lysates and culture supernatants concentrated by ultrafiltration using anti-HER2 antibody. (C) Soluble HER2 analyzed by ELISA in serum samples obtained from mice xenografted with HCC827 or NCI-H2170 cells.
Since immunohistochemistry using anti-HER2 has already been applied for determining Herceptin® treatment in breast cancer, it seemed to be a promising marker to predict HER2-addicted lung carcinomas. In the current study, anti-HER2 staining clearly distinguished the HER2-addicted cancer cells in the xenografted nude mice, and thus may be proven to be a powerful method to provide direct evidence of the HER2 status for lung carcinomas. However, immunohistochemistry requires excised tissue specimens, and would not be applicable for inoperable patients. Ectodomain shedding of HER2 has been reported and the soluble form of HER2, known to be secreted from several cancer cells including breast cancer (27) can be found in the serum of tumor bearing mice. Soluble HER2 was confirmed to be secreted into the culture supernatant only from the HER2-addicted NCI-H2170 cells. Furthermore, soluble HER2 was detected in vivo specifically in the serum of the mice xenografted with NCI-H2170 cells, suggesting that circulating soluble HER2 might be a promising, cost-effective and non-invasive marker to guide the appropriate treatment of patients with HER2-addicted lung carcinomas, using existing EGFR inhibitors or specific HER2 inhibitors which may be developed in the future. Further studies will be required to determine the clinical relevance of these findings.
Acknowledgements
We greatly thank H. Kurosawa for her technical assistance and other many colleagues in the Fundamental Research Department, Fujirebio Inc. for their support and discussions. This work was also supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology (20390082 and 1701468 to S. Higashiyama).
- Received August 31, 2011.
- Revision received October 27, 2011.
- Accepted October 28, 2011.
- Copyright© 2011 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- 3D genomic analysis reveals novel enhancer-hijacking caused by complex structural alterations that drive oncogene overexpression
- Tarloxotinib Is a Hypoxia-Activated Pan-HER Kinase Inhibitor Active Against a Broad Range of HER-Family Oncogenes
- 89Zr-Chloride Can Be Used for Immuno-PET Radiochemistry Without Loss of Antigen Reactivity In Vivo