


Abstract
The anti-tumor properties of arsenic trioxide have attracted extensive attention after successfully inducing apoptosis of acute promyelocytic leukemia cells. However, the therapeutic spectrum should not only be restricted to acute promyelocytic leukemia, but should also extend into other types of tumor cells. In this study, we aimed at investigating its potential application to clinical therapeutics in oral cancer. In this preclinical animal test, primarily cultured cells from the tumor sites and normal sites of a two-drug (200 μg/ml 4-nitroquinoline 1-oxide (4NQO) plus 500 μg/ml arecoline)-induced oral cancer C57BL/6J Narl mice model were examined for their viabilities after treatments of arsenic trioxide with/without other drugs. In this model, the mice were treated with 4NQO plus arecoline (NA) in their drinking water for eight weeks (8-w), and the drugs were withdrawn for another 10 or 20 weeks (18-w and 28-w, respectively). The results showed that 2 μM of arsenic trioxide 24-h treatment suppressed the viabilities of cells primarily cultured from the tumor sites of 8-w, 18-w and 28-w NA-treated mice to 72.9%, 71.5% and 65.6%. However, it also suppressed the viabilities of cells from the sham-treated mice of 8-w, 18-w and 28-w to 76.8%, 73.4% and 75.7%, respectively. Therefore, 0.5 μM of arsenic trioxide treatment for 24 h, which suppressed the viabilities of cells primarily cultured from the tumor sites of 28-w NA-treated and sham-treated mice to 15.6% and 9.1%, was examined for its synergistic effects on the two primarily cultured cell lines with other drugs. The results showed that 10-20 μM dithiothreitol enhanced the cytotoxic effects of arsenic trioxide to 43.3~62.1%, better than those of 4 J/m2 UVC, 20 μM H2O2 or 100 μM buthionine sulfoximine (21.3%, 13.2%, and 14.2%, respectively). At the same time, 10-20 μM dithiothreitol plus 0.5 μM arsenic trioxide treatments caused only 12.3% and 15.2% of cell death in the control group. The cytotoxicity of dithiothreitol and arsenic trioxide combination on primarily cultured cells from this oral cancer model should be confirmed in human oral cancer cell lines before its application in clinical therapy, and the detailed mechanism is worth further investigation.
Arsenic trioxide (As2O3), a traditional Chinese medicine renown for its toxicity, has been long used for medical purposes. It has been demonstrated to be effective for the treatment of acute promyelocytic leukemia (APL) (1, 2). As other useful anticancer drugs, it is very likely that As2O3 might be useful for the treatment of other types of cancer. Since the preclinical tests for any potential drugs are carefully performed both at the cell level and the animal model, we have tested the hypothesis of using As2O3 in oral cancer therapy in cells primarily cultured from a very well-established oral cancer mice model. In the model, male C57BL/6J Narl mice were treated with (NA groups) or without (sham-treated control group) 200 μg/ml 4-nitroquinoline 1-oxide (4NQO) plus 500 μg/ml arecoline in their routine drinking water for eight weeks, and then the drugs were removed for further twenty weeks. Immediately after the 8-week drug treatments (8-w), and each two weeks in the subsequent 20 weeks (10-, 12-, 14-, 16-, 18-, 20-, 22-, 24-, 26-, and 28-w), each of the mice were observed for the occurrence of oral cancer. Six mice from the NA group and six from the control group were randomly selected and sacrificed for tongue extraction and primary culture. The animal model has been shown to induce oral cancer with a 100% success rate after 28-weeks (3). In the literature, several compounds have been found to enhance the cytotoxicity of As2O3, including UVC (4), H2O2 (5), buthionine sulfoximine (6) and others (7, 8). In this study, the aim was to investigate the cytotoxic effects of As2O3 alone and As2O3 combined with other drugs, on the oral cancer cells extracted from the animal model.
Materials and Methods
Animals and drugs. Six-week-old male C57BL/6J Narl mice, purchased from the National Laboratory Animal Center (Taipei, Taiwan), were used in this study. The mice were handled in accordance with the Animal Care and Use Guidelines of the China Medical University, and the study protocol was approved by the Institutional Animal Care Use Committee. The experiments were carried out under controlled conditions with a 12-h light/dark cycle. Mice were fed standard mouse chow (Fwosow Industry Co. Ltd., Taiwan). All chemicals were purchased from Sigma (CNC Technology Machinery Co., Ltd., Taiwan). The carcinogens, 4-NQO (Sigma–Aldrich, St. Louis, MO, USA) and arecoline hydrobromide (Fluka, Buchs, China), were dissolved in the drinking water, which was replaced once a week. Mice were allowed to access the drinking water and chow diet ad libitum during the treatment. All mice were weighed every 4 weeks (3).
A novel model to induce tongue tumors by co-treatment of 4-NQO plus arecoline. Briefly, the co-treatment of the carcinogen 4-NQO (200 μg/ml) plus arecoline (500 μg/ml) in the drinking water for eight weeks to C57BL-6 mice specifically induced tongue cancer within a further 20 weeks, whereas no tumors occurred in the remainder of the digestive tract, lungs and liver. This model can induce oral cancer much more effectively (with shorter period and reasonable drug dosage) than any other model in the literature (9, 10).
Extraction of the tongues. Mice were sacrificed one by one, the whole tongue was carefully excised and images were taken before the sampling of tissues from the tumor sites and distal as possible from the tumor sites. The upper exteriors of the tongue from the tumor site and distal from the tumor (internal control) site of the 4-NQO plus arecoline co-treated (NA) mice, and the same sites from age-matched sham-treated mice (external control) were obtained from these mice (male, warm-blood) every two weeks from the end of the 8-week co-treatment. Immediately after their anesthetization, the whole tongue was dissected and steel scissors cuere used to cut the upper exteriors from the tumor site and distal (as far as possible from the tumor) site of the NA mice, and the same sites from age-matched sham-treated mice. Subsequently the piece of the tongue (approx. 5 to 10 mm long) was extracted and placed in DMEM containing 150 U/ml penicillin, 150 μg/ml streptomycin, and 10% fetal calf serum. All the above procedures were carried out at least three times and approved by the Institutional Review Board of Animal Committee of the China Medical University and Hospital. Isolation and preparation of primary cell culture from the upper exteriors of the tongue. The samples retrieved from the tongues were washed three times in DMEM with 10% fetal calf serum. After the final wash, the pieces were further cut with scissors into smaller pieces, and all suspensions were centrifuged (500 rpm, 5 min). The obtained pellets were incubated in DMEM containing 2% collagenease at 37°C for 15 min. Digestion was terminated by centrifugation (500 rpm, 5 min) and then re-suspended in culture medium. Subsequently, the cells were incubated in 25-mm flasks in a humidified atmosphere (95% air, 5% CO2, 37°C).
Measurement of cell viability. The sensitivity of cells to drugs was determined by a standard spectrophotometric 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) assay (Sigma-Aldrich) (11). Briefly, cells were seeded at a density of 105 cells/well in 100 μl into 96-well plates and incubated for 24 h at 37°C in an atmosphere of 95% air and 5% CO2. Cells grown for 24 h were then exposed to As2O3, or drug combination as indicated. After 24-h of drug treatment, the medium was aspirated, and the cells were rinsed once with PBS and serum-free culture medium without phenol red was replaced in all wells. Cells were then incubated for 4 h with MTT solution (5 mg/ml). The yellow tetrazolium salt was metabolized by viable cells to form purple crystals of formazan. The product was quantified spectrophotometrically by measuring absorbance at 570 nm using a microplate reader (Quant™, BioTek Instruments Inc., CA, USA). The cell viability was also cross checked by using Trypan blue assay and microscopic counting (12) and was expressed as a percentage of viable cells.
Statistical analysis. Results are expressed as the mean standard deviation. Differences between groups were determined by two-sided Student's t-test. Values of p<0.05 were considered statistically significant.
Results
The perfect animal model and primarily cultured cells. Over the years, NA-induced oral cancer in mice model has been repeated seven times and the 100% occurrence of oral cancer at week 28 (or even earlier) has been proven. The occurrence rates and appearances have also been similar among each round. Almost all of the tumors found were located on the tongue, and focused in the front sites and the central areas of the rear sites. Some of the mice had more than two primary tumor sites, and the largest number in one mouse was six. No buccal mucosa or other sites were found. The distribution percentages were very different from that in humans (about 35-40% were tongue and equal proportion were buccal mucosa), while the pathology was very similar (3).
The cytotoxicity of arsenic trioxide. Three time points have been chosen to represent the initiation (8-w), progression (18-w), and maturation (28-w) stages of oral carcinogenesis, to investigate the cytotoxicity of As2O3. The cells from sham-treated mice were the control groups. The dosage of As2O3 ranged from 0 to 8 μM and the treatment time was 24 h. The cytotoxicity of As2O3 to 8-w control, 18-w control, 28-w control, 8-w NA, 18-w NA, and 28-w NA were all dose-dependent (Figures 1, 2 and 3). Twenty-four hours with 2 μM As2O3 treatment suppressed the viabilities of 8-w, 18-w and 28-w NA-treated mouse tumor cells to 72.9%, 71.5% and 65.6%. However, it also suppressed the viabilities of cells from the sham-treated mice of 8-w, 18-w and 28-w to 76.8%, 73.4% and 75.7%, respectively. To a lesser extent, 24-h 0.5 μM of As2O3 treatment suppressed the viabilities of 8-w NA, 18-w NA, 28-w NA, 8-w control, 18-w control and 28-w control to 90.0%, 88.6%, 84.4%, 91.7%, 91.6%, and 90.9%, respectively (Figures 1, 2 and 3).
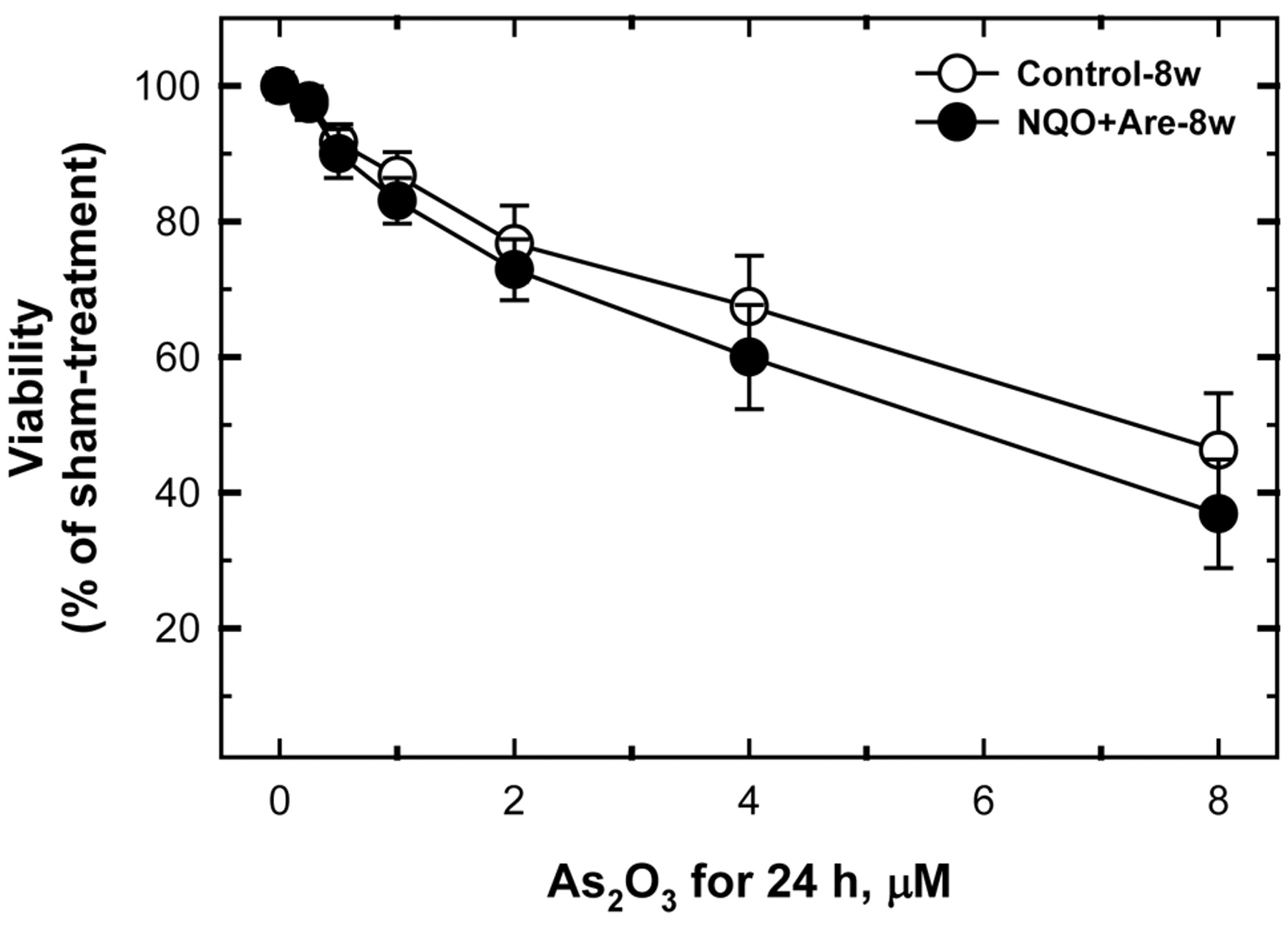
The dosage of arsenic trioxide ranged from 0 to 8 μM and the treatment time was 24 h. The 2 μM of arsenic trioxide 24-h treatment suppressed the viabilities of 8-w NA-treated mouse tumor cells to 72.9%. However, it also suppressed the viabilities of cells from the sham-treated mice of 8-w to 76.8%. To a lesser extent, 24h 0.5 μM of arsenic trioxide treatment suppressed the viabilities of 8-w NA NA-treated mouse tumor cells and 8-w control to 90.0% and 91.7%, respectively.
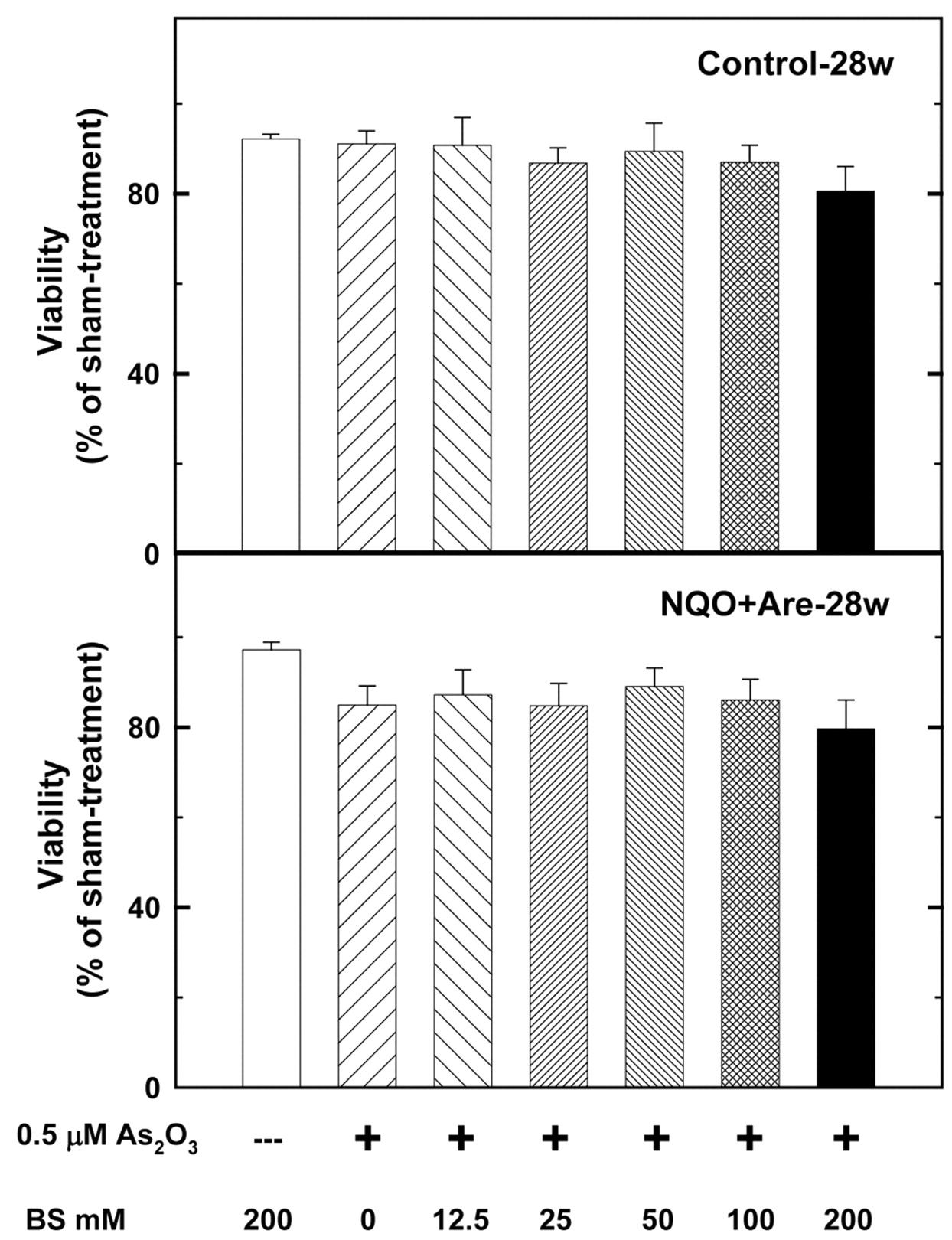
The cytotoxicity of As2O3 with UVC, H2O2, dithiothreitol, and buthionine sulfoximine. A 0.5 μM As2O3 treatment for 24 h, suppressed the viabilities of cells primarily cultured from the tumor sites of 28-w NA-treated and sham-treated mice by 15.6% and 9.1% (Figure 3) the treatment was examined for its synergistic influence on the two primarily cultured cell lines with other drugs. The results showed that 10 and 20 μM dithiothreitol can enhance the cytotoxic effects of As2O3 to 43.3 and 62.1%, respectively (Figure 4), better than 21.3% of 4 J/m2 UVC (Figure 5), 13.2% of 20 μM H2O2 (Figure 6) and 14.2% of 100 μM buthionine sulfoximine (Figure 7). At the same time, 10 and 20 μM dithiothreitol plus 0.5 μM As2O3 treatments caused only 12.3% and 15.2% of cell death in the control group (Figure 4).
Discussion
The anti-neoplastic properties of arsenic have aroused worldwide interest after it effectively induced apoptosis in APL cells (13, 14). The therapeutic spectrum should not only be restricted to APL, but may also extend to cells from other types of cancer (15, 16). However, the diverse sensitivity as in the less sensitive tumor cells requiring supra-therapeutic levels or up to tens of micromoles of arsenic (much higher than those used against APL) is an issue as that is not clinically achievable without the risk of arsenic-mediated side-effects (17). This poses a challenge for transitional medical scientists working on the search for ultra-low dose when using arsenic for any clinical therapy. The best solution for prevention of arsenic-induced toxicity is via the combinational or cock-tail access.

The dosage of arsenic trioxide ranged from 0 to 8 μM and the treatment time was 24 h. Arsenic trioxide at 2 μM for 24-h suppressed the viabilities of 18-w NA-treated mouse tumor cells to 71.5%. However, it also suppressed the viabilities of cells from the sham-treated mice of 18-w to 73.4%. To a lesser extent, 0.5 μM of arsenic trioxide 24-h treatment suppressed the viabilities of 18-w NA NA-treated mouse tumor cells and 18-w control to 88.6% and 91.6%, respectively.

The dosage of arsenic trioxide ranged from 0 to 8 μM and the treatment time was 24 h. Arsenic trioxide at 2 μM for 24-h treatment can suppressed the viabilities of 28-w NA-treated mouse tumor cells to 65.6%. However, it also suppressed the viabilities of cells from the sham-treated mice of 28-w to 75.7%. To a less extent, 0.5 μM of arsenic trioxide for 24-h suppressed the viabilities of 28-w NA NA-treated mouse tumor cells and 28-w control to 84.4% and 90.9%, respectively.

The dosage of dithiothreitol (DTT) ranged from 0 to 40 μM and the 0.5 μM arsenic trioxide treatment time was 24 h. Combination of 40 μM DTT and 0.5 μM arsenic trioxide suppressed the viabilities of 28-w NA-treated mouse tumor cells and sham-treated mice cells of 28-w to 38.0% and 85.0%. The synergistic effects for 20 μM DTT plus 0.5 μM arsenic trioxide for the two groups were statistically significant (p<1.0×10−7).
As a first step, we investigated the dose–response of oral cancer cells primarily cultured from the tumor sites of the well-established oral cancer mouse model to As2O3 treatment. Then the dose of 0.5 μM was chosen for further examinations of the potential agents. In the literature, several thiol-containing molecules (TCM) are commonly used as antidotes for arsenic, and vicinal TCM seem to be more effective in mobilizing tissue arsenic than are mono TCM. Interestingly, we have found that DTT, a typical vicinal TCM, has synergistic cytotoxic effects with As2O3 on oral cancer cells, while not on counterpart normal cells. In detail, 10 and 20 μM DTT can enhance the cytotoxic effects of As2O3 to 43.3 and 62.1%, respectively, much better than those of 4 J/m2 UVC, 20 μM H2O2 or 100 μM buthionine sulfoximine (21.3%, 13.2%, and 14.2%, respectively). Moreover, 10 and 20 μM DTT plus 0.5 μM As2O3 treatments caused only 12.3% and 15.2% of cell death in the control group.
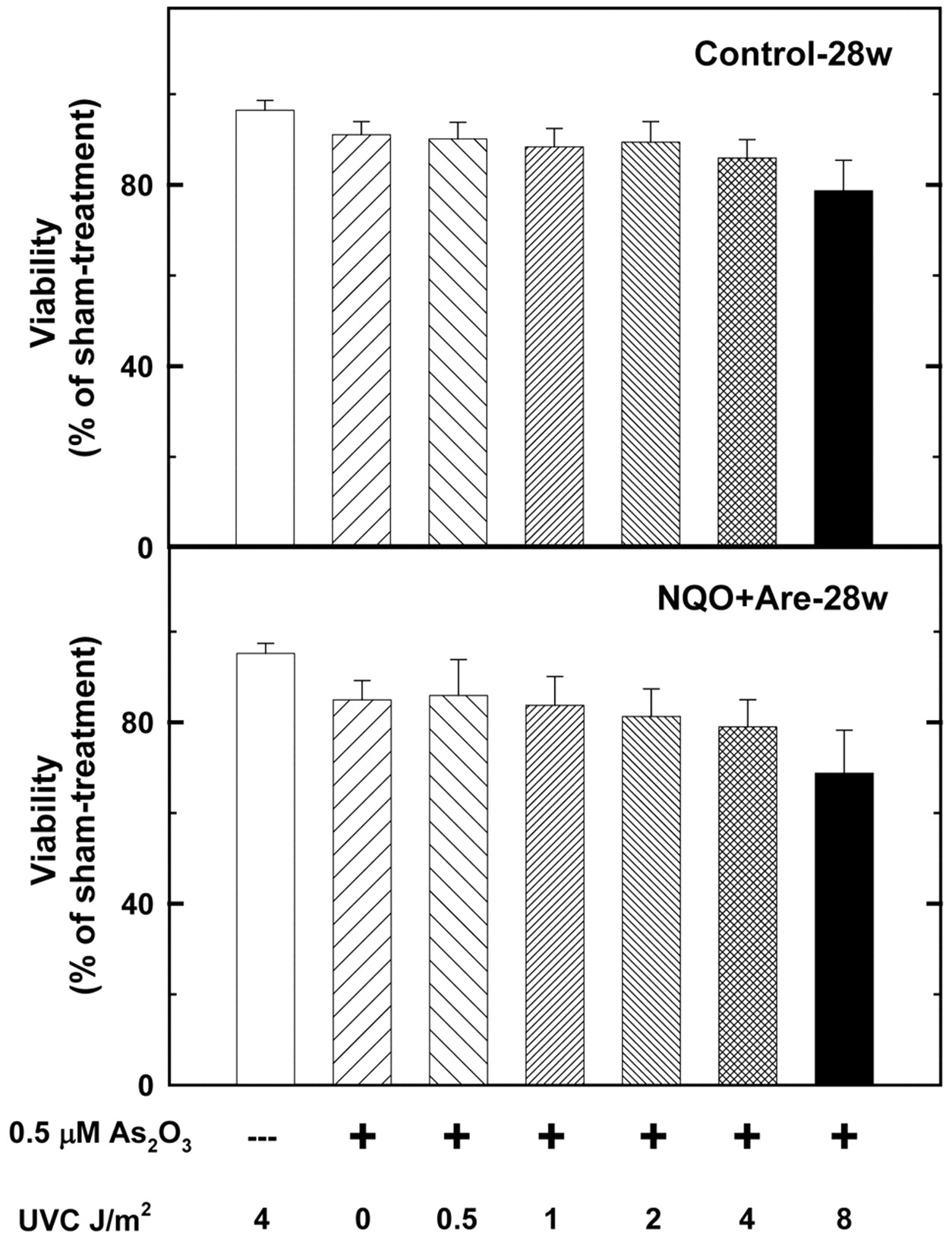
The dosage of UVC ranged from 0 to 8 J/m2 and the treatment time for arsenic trioxide at 0.5 μM was 24 h. 4 J/m2 UVC and 0.5 μM arsenic trioxide suppressed the viabilities of 28-w NA-treated mouse tumor cells and sham-treated mouse cells of 28-w to 79.0% and 85.9%. The synergistic effects of 4 J/m2 UVC plus 0.5 μM arsenic trioxide on the two groups were statistically significant (p=0.00025).
Whether DTT itself has cytotoxicis or cytotoxicity with its vicinal thiol groups, and whether other thiol-containing drugs can have similar or better effects than dithiothreitol was questioned. To investigate this hypothesis, we have collected as many investigate thiol-containing drugs as we could but the results were all negative (data not shown). It would be interesting to elucidate the co-cytotoxic mechanism of As2O3 plus DTT to oral cancer cells. However, based on the results obtained thus far, we can only exclude ‘thiol-binding’ as the main cause of the co-cytotoxicity. Arsenic may produce ROS and RNS (18-20) and NO can react with superoxide to produce peroxynitrite, which can cause oxidative DNA damage, nitrosylate cellular proteins and lipoproteins (21). ROS has been shown to be involved in arsenite-induced DNA damage (22, 23), gene mutation (24), cell proliferation (25), and apoptosis (26, 27), while RNS has been shown to be involved in DNA repair inhibition (19, 28). Each of the outcomes investigated plays a role in the cytotoxicity. Although data are missing to reveal the effects of DTT alone and As2O3 plus DTT on oral cancer cells, the virtue of finding the co-cytotoxicity is valuable for potential clinical applications just as in the case of As2O3 for APL.
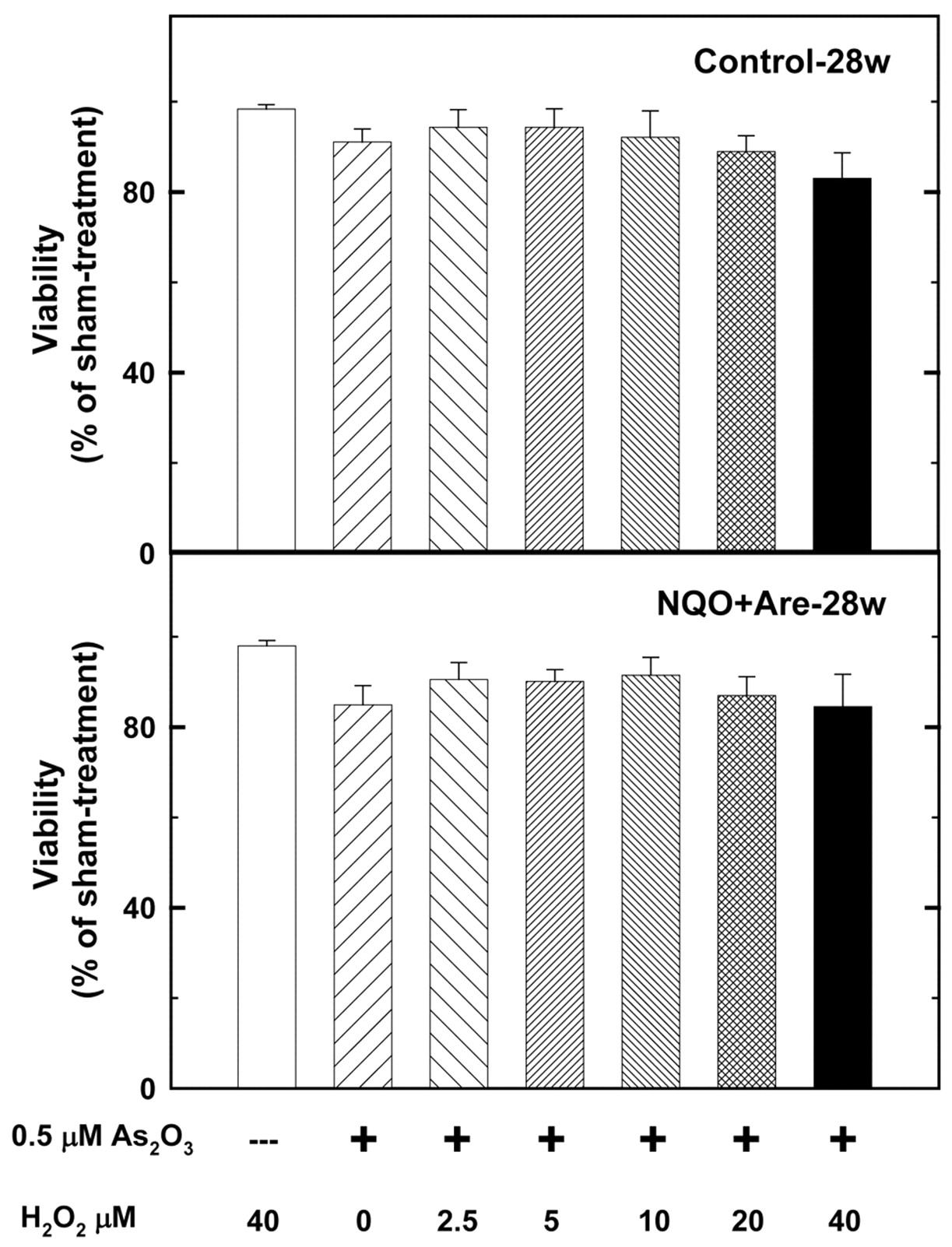
The dosage of H2O2 ranged from 0 to 40 J/m2 and 0.5 μM arsenic trioxide treatment time was 24 h. 40 μM H2O2 and 0.5 μM arsenic trioxide suppressed the viabilities of 28-w NA-treated mouse tumor cells and sham-treated mouse cells of 28-w to 87.0% and 89.0%. The synergistic effects of 40 μM H2O2 plus 0.5 μM arsenic trioxide on the two groups were not statistically significant (p=0.1299).
Elucidation of the detailed mechanism is worth further investigations. One approach could involve the measurements of ROS and RNS produced by the drugs (alone or combined), in an attempt to modulate them and investigate influences on the end-points. The co-cytotoxicity of DTT and As2O3 on primarily cultured cells from oral cancer mice also needs to be confirmed in oral cancer cell lines from oral cancer patients, prior to its application in clinical therapy. Pre-clinically, the co-cytotoxicity of As2O3 plus DTT is specific to oral cancer cells but not to the normal cells primarily cultured from a perfect oral cancer cell model. This novel finding needs to be confirmed in oral cancer cells from human patients, and the detailed mechanism pursued through further studies.

The dosage of buthionine sulfoximine (BS) ranged from 0 to 200 mM and the 0.5 μM arsenic trioxide treatment time was 24 h. Combination of 200 mM BS and 0.5 μM arsenic trioxide suppressed the viabilities of 28-w NA-treated mouse tumor cells and sham-treated mouse cells of 28-w to 86.1% and 87.0%. The synergistic effects of 200 mM BS plus 0.5 μM arsenic trioxide on the two groups were not statistically significant (p=0.5070).
Acknowledgements
We thank Wen-Shin Chang and the Tissue Bank at the China Medical University for their technical assistance. This study was supported by research grants from the China Medical University and Hospital (DMR-99-045), National Science Council (NSC 98-2320-B-039-010-MY3), and Taiwan Department of Health, China Medical University Hospital Cancer Research of Excellence (DOH99-TD-C-111-005).
- Received May 26, 2010.
- Revision received June 22, 2010.
- Accepted June 28, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.