


Abstract
Aim: To identify the effect of a TGF-β1 antisense treatment of keloid fibroblasts on the SMAD signalling system. Material and methods: In this cross-sectional study, keloid and adjacent healthy tissue was harvested from 9 patients with keloid scars after otoplasty. Keloid fibroblasts were placed in monolayer cultures. Expression of SMAD2, -3, -4, -6, and SMURF2 were analysed by immunohistochemistry. Analysis of treatment with antisense oligonucleotides was conducted by immunohistochemistry, and RT-PCR. Results: Immunohistochemical investigation demonstrated increased expression of SMAD2, -3 and -4, and decreased expression of SMURF2. TGF-β1 antisense therapy significantly down-regulated SMAD2 and SMAD4, up-regulated SMURF2 and showed no effect on SMAD3 and SMAD6. Conclusion: TGF-β1 led to elevated levels of the SMAD signalling cascade, indicating an abnormal sensitivity of keloid-derived fibroblasts to this cytokine. Abrogation correlated with potential suppression of the fibro-proliferative progress. There is growing evidence for an abnormal response to this cytokine in the intracellular signal transduction in keloid-derived fibroblasts.
Keloids represent one extreme of aberrant dermal wound healing observed in susceptible individuals after cutaneous injury, creating unsightly scars that contain atypical fibroblasts and an overabundance of extracellular matrix components (ECM), in particular collagen, fibronectin and certain proteoglycans (1, 2). Clinically keloids extend beyond the boundaries of the original wound and grow continuously, but do not expand beyond the dermis (in contrast to malignant tumours) and are thus benign lesions (3). Surgical excision with intralesional steroid injection is a standard method of treatment. However, recurrence is commonly observed after surgical therapy, often exacerbating the primary condition. So far no satisfactory treatment exists (4, 5). These clinical findings reflect the scarce knowledge of the exact pathogenetic mechanism of these lesions, and the exact biomolecular mechanisms promoting keloid formation remain to be elucidated. A variety of explanatory approaches or theories of keloid pathogenesis have led to a multitude of studies. Consistent among these theories is the observation of excessive collagen deposition secondary to cutaneous trauma or injury, based on an abnormal collagen metabolism of keloid fibroblasts (1, 2, 6, 7).
More recently research has been focussed on the biomolecular pathways responsible for excessive ECM accumulation. Altered signal transduction or changes of regulatory pathways are currently proposed as being responsible for the increased content of ECM material in keloids (8). Transforming growth factor β1 (TGF-β1) is well known as a crucial fibrogenic cytokine promoting ECM production and tissue fibrosis. There is emerging consensus that this cytokine plays a key role in keloid pathogenesis (1, 9-12). All three isoforms of TGF-β exert their influence by binding to the TGF-β type I and type II (TGF-βRI and TGF-βRII) receptors (11). Activation of these receptors by linkage with TGF-β leads to the transmission of a signal from the cell surface to the nucleus through interactions with downstream effector molecules, mainly including members of the SMAD family. SMAD proteins, the first proteins to be identified by TGF-βRI, play a central role in the transduction of receptor signal to target genes in the nucleus (6, 13). Three major groups of SMADs have been identified: in the presence of TGF-β ligand, the receptor-activated SMADs (R-SMADs), SMAD2 and SMAD3, are phosphorylated directly by the TGF-βRI kinase, bind to the common mediator SMAD (Co-SMAD), SMAD4, and translocate into the nucleus. Besides R-SMADs and Co-SMAD, a third group of SMADs, the inhibitory SMADs (I-SMAD) such as SMAD6, prevent R-SMAD phosphorylation and subsequent nuclear transduction of R-SMAD/SMAD4 heterocomplexes. I-SMADs exert a negative feedback by competing with R-SMADs for receptor interaction and by marking the receptors for degradation (14, 15). Figure 1 illustrates the functional correlations between the different members of the SMAD signaling cascade within human fibroblasts.
It has been reported that TGF-βRI, TGF-βRII and SMAD proteins are highly expressed in keloid fibroblasts compared to normal skin fibroblasts, indicating that keloid formation may be caused by up-regulation of the TGF-β/SMAD signalling pathway (13, 16, 17). The key to understanding the pathogenesis of keloid formation may rest on understanding the impact of TGF-β1 on different functional groups of wound healing. In previous studies the correlation of TGF-β1 with its isoforms or receptors, or with relevant integrins in keloid-derived fibroblast was analysed (12, 18). The aim of this study was to investigate the effect of TGF-β1 targeting by antisense oligonucleotides on the SMAD signalling system in keloid-derived fibroblasts.
Materials and Methods
Immunohistochemistry. Tissue specimens from 9 patients with keloid scars after otoplasty and control biopsies of normal healthy tissue (from the same patient) were obtained during surgery, which was performed at the Department of Otolaryngology, Head and Neck Surgery, University Hospital of Mannheim. The study protocol was approved by the ethics committee of the institution, and written consent was obtained from all subjects for all procedures. A sample from each resected scar was sent to the pathology laboratory for histologic processing and confirmation of the clinical diagnosis. Samples were frozen in liquid nitrogen for SMAD identification. For in vitro analysis dermal fibroblasts isolated from keloids and normal controls were cultured in Falcon petri dishes (Greiner, Germany) at 37°C in a 5% CO2 fully humidified atmosphere in serum-free Fibroblast Growth Medium (PromoCell, Heidelberg, Germany) supplemented with antibiotics (Gibco BRL Life Technologies Inc., Gainthersburg, MD, USA). The immunohistochemistry for SMAD2, SMAD3, SMAD4, and SMURF2 was performed using the streptavidin-biotin complex procedure (Amersham, Braunschweig, Germany). After blockage of endogenous peroxidase with 0.3% hydrogen peroxidase for 30 min the sections were washed with phosphate-buffered saline (PBS) and incubated with normal rabbit serum in PBS for 30 min at room temperature in order to block non-specific antibody reaction. Sections were then incubated overnight at 4°C with the primary antibody. The slides were washed in several changes of PBS and then incubated with a peroxidase-conjugated secondary antibody (DAKO, Hamburg, Germany). After being washed twice in PBS, the sections were treated with a streptavidin-biotin-peroxidase complex and peroxidase reaction was performed using Diaminobenzidine DAB (DAKO) as chromogen. Different antibodies were diluted to the desired concentrations in PBS. Controls were carried out by omitting the primary antibody. Light microscopical investigation was performed using a Zeiss Axiophot microscope (Zeiss, Oberkochen, Germany).
Oligodeoxynucleotides. Phosphorithiotated 14-mer oligodeoxynucleotides (ODN) were synthesised on an Applied Biosystems 394 DNA synthesiser (Applied Biosystems Inc., Foster City, CA, USA) by means of B-cyanothylphosphoramidite chemistry to minimise degradation by endogenous nucleases. The antisense oligonucleotide (5′-CGA TAG TCT TGC AG-3′) was directed against the translation start site and surrounding nucleotides of the human TGF-β1 cDNA. The in vitro inhibitory effect of these antisense ODNs on TGF-β1 expression at both the mRNA and protein level in human cells had been described previously (19). All experiments were performed with 3 μM ODNs. To determine the effect of oligonucleotides on the expression of SMAD mRNAS or SMURF2 mRNA, fibroblasts were plated at a density of 105 cells/microtiter well in 24 well polystrene plates (Falcon; Becton Dickinson Labware, Franklin Lakes, NJ, USA). After 24 hours, the cells were rinsed twice with medium and then fresh TGF-β1 oligo medium containing antisense ODNs was added, followed by an incubation period of 48 h.
RT-PCR. To isolate the RNA from the fibroblasts grown in monolayer, the cells were directly lysed in the culture dish by addition of 1 ml RNA-Clean (RNA-Clean System, AGS, Heidelberg, Germany). After addition of 0.2 ml chloroform per 2 ml of homogenate and centrifugation for 15 minutes at 12,000 ×g (4°C), the supernatant was removed from the RNA precipitate. The RNA pellet was washed twice with 70% ethanol by vortexing and subsequent centrifugation for 8 minutes at 7,500 ×g (4°C). After drying the RNA pellet, it was dissolved in DEPC water. The RNA was reverse transcribed (StrataScript First-Strand Synthesis System, Stratagene, La Jolla, CA, USA) into cDNA using random-oligunucleotide primers. Primer sequences used for RT-PCR were as follows: for SMURF2: sense 5′-GTT GTG ATG GGT TCT GAT TC-3′ and antisense 5′-CAC CAA TGG CAA AAG GCT-3′; for SMAD2: sense 5′-GGA GCA GAA TAC CGA AGG CA-3′ and antisense 5′-CTT GAG CAA CGC ACT GAA GG-3′; for SMAD3: sense 5′-AGA AGA CGG GGC AGC TGG AC-3′ and antisense 5′-GAC ATC GGA TTC GGG GAT AG-3′; for SMAD4: sense 5′-CAT CGA CAG AGA CAT ACA G-3′ and antisense 5′-CAA CAG TAA CAA TAG GGC AG-3′ and for SMAD6: sense 5′ CAA GCC ACT GGA TCT GTC CGA-3′ and antisense 5′-TTG CTG AGC AGG ATG CCG AAG-3′. SMAD and SMURF2 mRNA levels were measured in all cell types using RT-PCR (MMP-CytoXpress Multiplex PCR Kit, Bio Source, San Francisco, CA, USA) according to the manufacturer's instruction manual. To fractionate the MPCR DNA products, the MPCR products were mixed with 6× loading buffer and separated on a 2% agarose gel containing 0.5 mg/ml ethidium bromide, visualised with UV light and recorded using a CCD camera. To test the quality of the cDNA, the kit included primers for GAPDH. Results were obtained in two independent experiments.
Results
Immunohistochemical in vitro investigation demonstrated an increased expression of SMAD2, SMAD3, and SMAD4 in all keloid tissues in comparison to normal human skin controls. SMURF2 was decreased in keloid fibroblasts (data not shown).
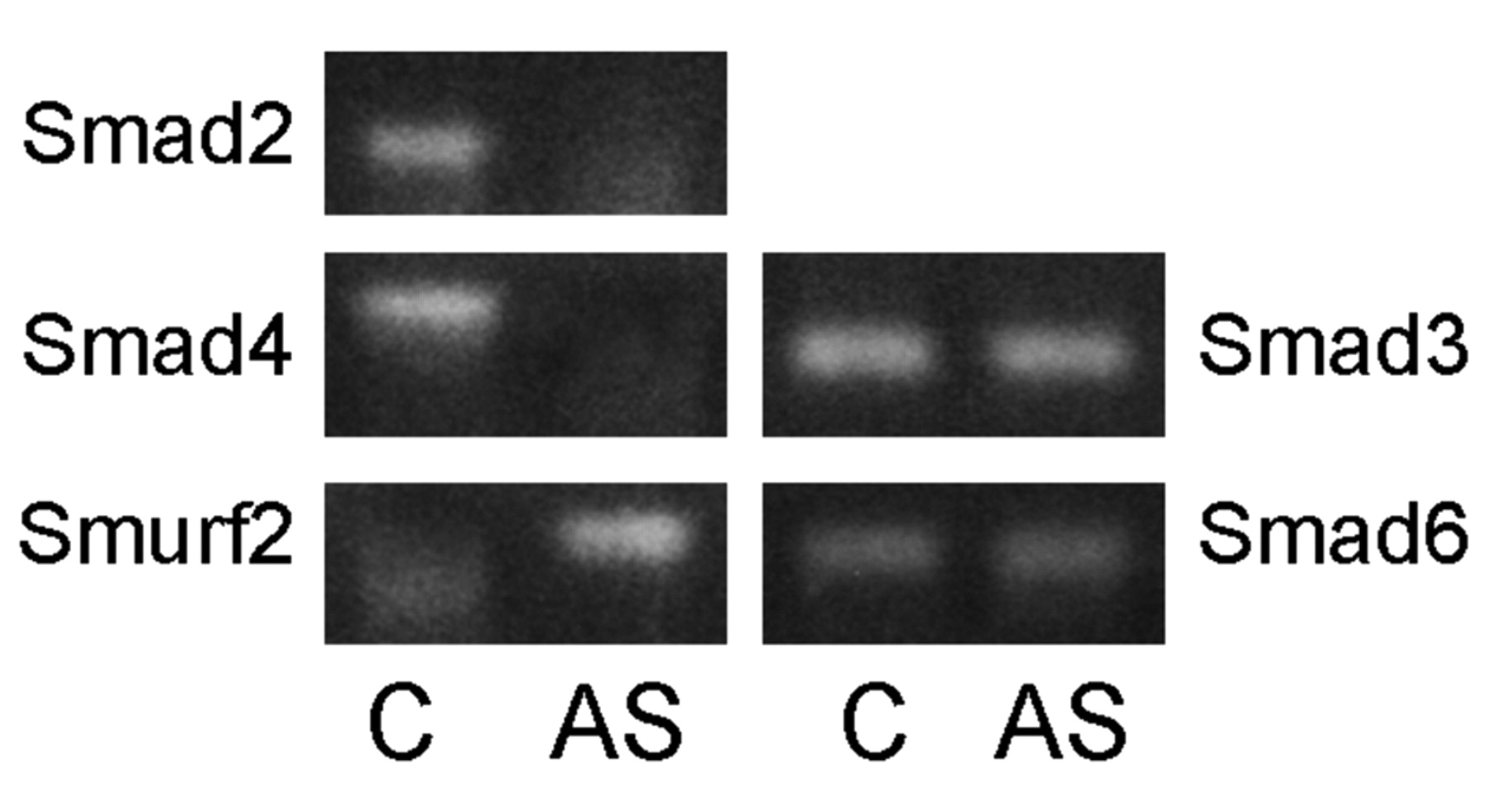
After treatment of keloid fibroblasts with TGF-β1 antisense oligodeoxynucleotides (ODNs) in vitro, expression of mRNA of SMAD2, SMAD3, SMAD4, SMAD6 and SMURF2 was measured using the multiplex RT-PCR kit. Incubation time was 48 h containing 3 μM TGF-β1 antisense ODNs. Expression patterns of SMAD2 and SMAD4 were decreased after antisense treatment. Expression of SMURF2 mRNA was up-regulated. SMAD3 and SMAD6 mRNA expression did not change significantly after addition of TGF-β1 antisense ODNs (Figure 2).

Model showing the TGF-β/SMAD signalling system. TGF-β1 binds to TGF-βRII. Once the ligand is bound, TGF-βRII binds to TGF-βRI forming a complex. The TGF-β receptor kinase phosphorylates the receptor-regulated SMADs (SMAD2/3) which then activate SMAD4 (the common partner SMAD), forming a heterocomplex, which translocates to the nucleus. The negative feedback mechanisms regulate the signal transduction in two ways: either by competitive inhibition of R-SMAD phosphorylation by inhibitory SMADs (e.g. SMAD6 or SMAD7) or by inhibition of formation of the heterocomplex by SMURF2.

Expression of mRNA in keloid fibroblasts before C and after 48 hours of treatment with TGF-β1 antisense ODNs (AS). Left column: significant changes in expression patterns of SMAD2, SMAD4 and SMURF2 mRNAs; right column: no changes in the expression of SMAD3 mRNA and SMAD6 mRNAs.
The changes described for the mRNA level were also demonstrated after immunohistochemical investigation. Treatment with TGF-β1 antisense ODNs revealed a decreased expression of SMAD2 and SMAD4. The expression of SMAD3 did not change after incubation with the antisense. Immunohistochemical staining revealed increased expression levels for SMURF2 (Figure 3).
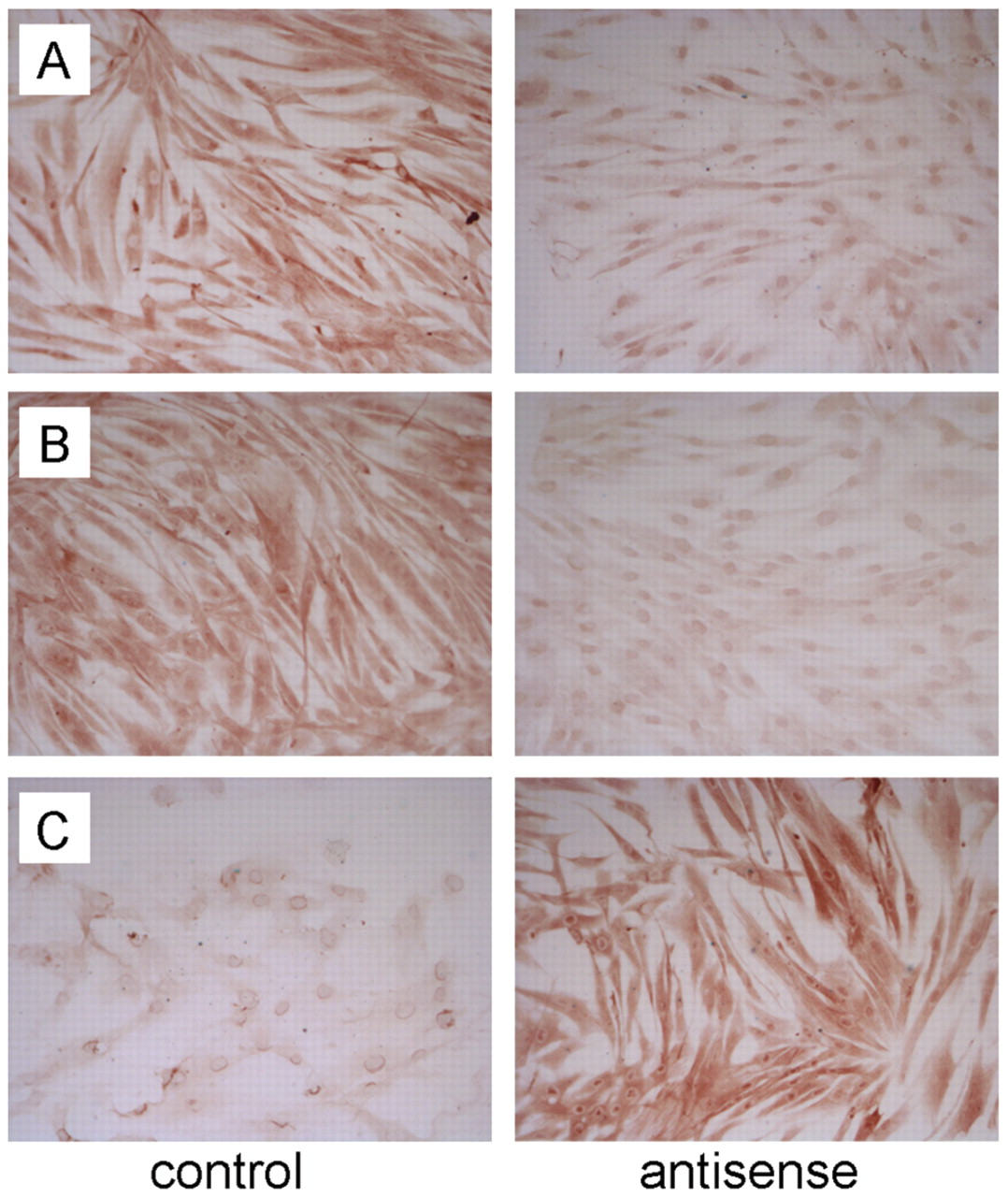
Immunohistochemical staining of keloid fibroblasts after treatment with TGF-β1 antisense ODNs. A: Expression of SMAD2, B: expression of SMAD4, and C: expression of SMURF2, before (control) and after treatment with TGF-β1 antisense ODNs (Magnification ×250).
Discussion
In order to ameliorate the understanding of biomolecular correlations of TGF-β1 in keloid scar tissue, this study investigated the effect of abrogation of TGF-β1 on the TGF-β/SMAD signal transduction system in keloid derived fibroblasts. The expression of SMAD after abrogation of TGF-β1 was analysed both at the transcriptional and the posttranscriptional levels. The findings demonstrate an increased activity of the SMAD signalling system within keloid fibroblasts. In a previous study, a correlation between targeting of TGF-β1 by antisense ODNs and the TGF-β-isoforms and their specific receptors was identified (12). The levels of profibrotic TGF-β-isoforms and TGF-β-specific receptors were increased in keloid fibroblasts. Therefore, it appears to be a logical consequence that elevated levels of TGF-β-ligands and receptors lead to excessive TGF-β-signalling (20). The data of the present study support this hypothesis, since there were elevated levels of SMAD2, SMAD3 and SMAD4, confirming the abnormal sensitivity of keloid-derived fibroblasts to TGF-β1, as reported elsewhere (9, 10). Increased levels of these SMADs, may lead to an increased signal transduction resulting in an overabundance of extracellular matrix components. Xie et al. observed an increased expression of SMAD2 after stimulation with TGF-β1 in hypertrophic scar fibroblasts and hypothesized that elevated levels of SMAD2 would make these cells more responsive to TGF-β1 (15). The data of the present study substantiated the fact that excessive keloidal scarring is associated with SMAD2 overexpression, accompanied by an enhanced activation of the associated SMADs of the intracellular signalling cascade.
Chin et al. documented a correlation between the TGF-β receptors and the downstream signalling molecule SMAD3 (16). Mice lacking Smad3 showed accelerated wound healing and reduced local inflammatory response (21). However, after abrogation of TGF-β1 in the present study, there was no corresponding effect on the transcriptional or posttranscriptional levels within keloid fibroblasts. This finding may imply an abnormal response to TGF-β1 in keloid fibroblasts by means of an altered intracellular signal transduction. Another indication for this hypothesis could be observed in the group of the inhibitory proteins SMAD6 and SMURF2. Both are well known for their inhibitory function forming intracellular complexes and thereby preventing translocation of the SMAD-complex to the nucleus (22, 23). Therefore, expression of these inhibitory proteins influences the TGF-β transcriptional responsiveness. The greater their expression levels are, the higher the level of resistance to profibrotic reactions will be (15, 24). In the present study, targeting of TGF-β1 up-regulated the expression of SMURF2 only, with no effect on SMAD6 mRNA levels. These observations suggest that a disruption in the SMAD6 pathway may be involved in keloid pathogenesis. This finding could also indicate an abnormal response to TGF-β1-signalling; however, further studies are needed to investigate these findings.
In conclusion, these results substantiate the notion that elevated levels of TGF-β1 lead to an up-regulation of TGF-β related signalling components, thus, implying a hyperactivity of the downstream intracellular molecules. Decreased expression of inhibitory components could be a reason for continuous growth in keloids due to a missing auto-negative feedback loop. Furthermore, these observations strengthen the paradigm that keloid pathogenesis may be the result of an aberrant intracellular signalling response.
Acknowledgements
The Authors would like to thank Petra Prohaska, Department of Otorhinolaryngology, Mannheim, for her excellent technical assistance.
- Received March 28, 2010.
- Revision received May 21, 2010.
- Accepted May 27, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.