


Abstract
Human tissue kallikrein 7 (hK7), a chymotrypsin-like secreted serine protease, catalyzes the degradation of intercellular adhesive structures in the cornified layer of the skin, leading to desquamation. Thus, hK7 is implicated in cancer invasion and metastasis. Although hK7 is highly expressed in prostate tissues, its biological role in prostate cancer progression is poorly understood. In the current study, we established an hK7-expressing cell model for prostate tumors by stably transfecting prostate carcinoma 22RV1 and DU145 cells with an expression vector encoding hK7. We found that there were no obvious differences in cell proliferation between cells overexpressing hK7 and cells transfected with empty vector (p>0.05). Intriguingly, a Matrigel invasion assay revealed that hK7 remarkably increased the migration and invasion of prostate cancer cells (p<0.01). Furthermore, hK7 induced epithelial−mesenchymal transition-like changes in prostate carcinoma cells, as evidenced by scattered cellular growth, mesenchyma-like morphology, and up-regulated expression of vimentin, a mesenchymal marker. These novel findings suggest that hK7 plays an important role in mediating prostate cancer progression and that hK7 promotes invasion and metastasis, at least in part, through inducing the epithelial−mesenchymal transition of prostatic carcinoma cells.
Human tissue kallikreins (hKs), encoded by the largest contiguous cluster of protease genes located on chromosome 19q13.4, are secreted serine proteases with diverse expression patterns and physiological roles. In addition to their well-known applicability as biomarkers (e.g. PSA/hK3, hK2, hK6, hK10, and hK11) in various types of cancer, certain hKs are implicated in many cancer-related processes such as invasion, proliferative regulation, and angiogenesis, by acting individually or in cascades with other hKs or proteases (1). The human tissue kallikrein hK7, encoded by the KLK7 gene, was initially purified from extracts of human skin (2). Subsequent studies have identified hK7 as a chymotrypsin-like serine protease that plays a central role in the turnover of the stratum corneum in the skin, by degrading intercellular adhesive structures and shedding epidermal cells; owing to these activities, hK7 has been implicated in stratum corneal diseases (3). Recent studies have suggested that hK7 is also associated with poor prognosis and progression of cancer. The presence of hK7 in serum has been proposed as an unfavorable marker in patients with cancer of the ovary, breast, and uterine cervix. Furthermore, overexpression of hK7 leads to the development of aggressiveness in ovarian, intracranial, and pancreatic cancer cells. However, there are few reports concerning a potential role of hK7 in prostate cancer progression.
Using an immunofluorometric assay, Kishi and colleagues have identified relatively high levels of hK7 in prostate tissue extracts and seminal fluid (4). Accordingly, in our previous studies, we have confirmed that hK7 is expressed by epithelial cells in the prostate and that the expression is up-regulated by certain paracrine factors derived from prostate stromal cells, suggesting that hK7 may function via stromal−epithelial interactions to affect pathological processes in the prostate.
The present study was undertaken to assess the role of hK7 in prostate cancer progression. We established a cell model for prostate tumors by transfecting prostate carcinoma 22RV1 and DU145 cells with an expression vector encoding hK7. Using this model system, we investigated whether enforced hK7 overexpression affects malignancy-related phenotypes in vitro.
Materials and Methods
Cell lines and cell culture. The prostate cancer cell lines 22RV1 and DU145 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA), 100 units/ml penicillin, and 100 mg/ml streptomycin, at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Transfection and selection. The coding sequence of KLK7 cDNA, amplified using mRNA extracted from normal human primary prostate epithelial cells as template and specific primers (sense: 5′-ACCATGGCAAGATCCCTTCTCC-3′; antisense: 5′-TTAGCGAT GCTTTTTCATGGTGTC-3′), was inserted into the mammalian expression vector pcDNA3.1 (Invitrogen) to construct the recombinant plasmid pcDNA3.1-KLK7. Sequencing was performed to verify the amplicon, and pcDNA3.1-KLK7 was transfected into DU145 and 22RV1 cells using Lipofectamine™ 2000 reagent (Invitrogen) according to the manufacturer's instructions. At 48 h after transfection, selective medium containing G418 antibiotic (Invitrogen) was added to the transfected cell cultures, at a final G418 concentration of 450 μg/ml for 22RV1 cells and 1000 μg/ml for DU145 cells. After 4 weeks in selective medium, stable transfectants of 22RV1 and DU145 grew into visible colonies. Eleven of the 22RV1 neo-resistant transfectants were randomly picked and grown separately in selective medium. Three of these clones were successfully expanded and prepared for further experiments. We designated the 22RV1 clones as RV-KL, RV-KM, and RV-KH based on their low, middle, and high KLK7 transcriptional levels, respectively, as detected by quantitative real-time RT-PCR. We pooled all of the KLK7-transfected DU145 clones and denoted the cell population as DU-K. For hK7-negative controls, 22RV1 and DU145 cells were also transfected with empty pcDNA3.1 vector and were selected using G418; the neo-resistant clones were mixed and designated as RV-con and DU-con, respectively. The levels of KLK7 transcription in these clones were determined by quantitative real-time RT-PCR with KLK7-specific primers (sense: 5′-AAGCCCAGGGTGACAAGA-3′; antisense: 5′-GTCGCCCAGCGTATCACT-3′).
Preparation of conditioned medium. For further confirmation of hK7 expression in subsequent experiments, conditioned medium (CM) of 22RV1 transfectants was prepared. Cells were detached by treatment with 0.25% trypsin-EDTA (Invitrogen), suspended in RPMI medium with 10% FBS, and seeded on 60-mm cell culture dishes. After a 24-h incubation for cell attachment, cultures at approximately 80% confluence were rinsed twice in phosphate buffered saline (PBS) and further cultured in 6 ml of serum-free RPMI-1640 medium at 37°C for 36 h. The supernatant was collected, cellular debris was removed by centrifugation at 300 ×g for 10 min, and the resulting CM was stored at −80°C. The cells were detached from the dishes, suspended, and counted using a hemacytometer. The ratio of CM volume to cell number was calculated to determine the relative ‘concentration’ of CM and the amount to be analyzed on Western blots.
RNA isolation, RT-PCR, and quantitative real-time RT-PCR. Total RNA was isolated from cells using an RNeasy Mini kit (Qiagen, Valencia, CA, USA), and mRNA was reverse transcribed with a Revert Aid™ First-Strand cDNA synthesis kit (Fermentas, MD, USA) according to the manufacturer's instructions. RT-PCR and real-time PCR of the cDNA were performed with primers specific to the genes of interest (KLK7 F: 5′-AAGCCCAGGGTGACAAGA-3′, R: 5′-GTCGCCCAGCGTATCACT-3′; 191 bp; vimentin F: 5′-AATGGCTCGTCACCTTCG-3′, R: 5′-AGTTTCGTTGATAACC TGTCC-3′; 249 bp). The RT-PCR products were separated by electrophoresis in 1.5% agarose gels, and the bands were visualized with UV light and photographed. SYBR Green I-based quantitative real-time PCR was carried out on a continuous fluorescence detection system (Opticon Monitor II; MJ Research, Inc., Watertown, MA, USA). Data were analyzed using the comparative Ct method (5). A difference of 1 between Ct values represents a 2-fold difference in the level of mRNA. The specificity of the PCR products was confirmed by melting curve analysis. The levels of gene expression were normalized to the expression of the housekeeping gene hypoxanthine-guanine phosphoribosyl transferase (HPRT; F: 5′-TGACACTGGCAAAACAATGCA-3′; R: 5′-GGTCCTTTTCACCAGCAAGCT-3′; 94 bp).
Western blot analysis. For Western blot analysis, samples were separated in 10% SDS polyacrylamide gels under denaturing conditions. The separated proteins were electroblotted onto Immobilon-P membranes (PVDF; Millipore Corporation, Bedford, MA, USA), and the membranes were blocked overnight at 4°C with 4% skim milk. The membranes were probed with the following as primary antibodies: rabbit anti-hK7 polyclonal antibody (1:500 dilution; Abcam, Cambridge, UK), mouse anti-vimentin monoclonal antibody, and mouse anti-β-actin monoclonal antibody (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA). After incubation with the primary antibodies, the membranes were incubated for 1 h at room temperature with horseradish peroxidase-conjugated goat-anti-rabbit (for hK7) or goat-anti-mouse (for vimentin and β-actin) secondary antibodies (Santa Cruz). The immunoreactive bands were visualized using an enhanced chemiluminescence reagent (Pierce, Rockford, IL, USA) and exposure to photographic film, according to the manufacturer's instructions.
MTT cell proliferation assay and colony formation assay. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) proliferation assays were based on an established method (6) with modifications. The 22RV1 transfectants were seeded at a density of 10,000 cells/well, and DU145 transfectants (DU-K and DU-con) were seeded at 5,000 cells/well in 96-well culture plates. All assays were performed with four replicates every 24 h, for a total of 96 h. Experiments were repeated three times. Data at 96 h were statistically analyzed by one-way ANOVA. For the colony formation assay, DU-K and DU-con cells were sparsely seeded at 500 cells/well in 6-well culture plates or at 3,000 cells per 60-mm culture dish. On day 15, when colonies were visible, the cell colonies were fixed in methanol, stained with 0.1% crystal violet, and photographed under a microscope.
In vitro Matrigel invasion assay. Matrigel (BD Biosciences, Bedford, MA, USA) is a solubilized basement membrane from Engelbreth-Holm-Swarm mouse sarcoma. The invasion of DU145 cells through 8-μm pores was assessed using Matrigel-coated Millicell culture plate inserts (Millicell-RCF; Millipore) according to the manufacturer's instructions. Viable cells (2×105) were seeded in the upper chamber with 400 μl of serum-free DMEM, and 600 μl of complete DMEM containing 10% FBS were placed in the lower chamber as a chemoattractant. After incubation at 37°C for 36 h, the medium was discarded, and the cells were washed twice with warm PBS. The cells and Matrigel in the upper chamber were removed using a cotton swab, and the cells that had migrated to the underside were fixed with 100% methanol for 30 min at 4°C. The methanol was aspirated, the cells were stained with 0.1% crystal violet solution at room temperature for 10 min, and excess dye was removed by rinsing with distilled water. The number of invading cells was counted under a microscope. The experiment was repeated three times, and statistical analysis was performed by one-way ANOVA.

Detection of hK7 protein in conditioned medium (CM) by Western blot analysis. Appropriate amounts of CMs produced by the transfectants (8 μl of 22RV1 transfectants; 18 μl of DU145 transfectants) were loaded into the lanes according to the ratio of the CM volume to the cell number, such that the CM in each lane was produced by an equal number of cells for 36 h, and the band intensity reflected the expression level of hK7 in each transfectant. The lanes loaded with CM from RV-KL, RV-KM, RV-KH, and DU-K cells show a specific band at about 28 kDa, with different intensities indicating the different hK7 concentrations in the CMs. No bands were recognized by the anti-hK7 antibody in the CM from RV-con or DU-con cells.
Treatment of wild-type DU145 cells with conditioned medium (CM) containing hK7. Wild-type DU145 cells maintained in RPMI-1640 medium were detached, counted, and seeded at approximate 1×105 cells/well on 24-well plates (Corning, NY, USA). After 36 h of incubation at 37°C for attachment, the culture medium in each well was replaced with 400 μl of serum-free RPMI-1640 and 200 μl of CM from 22RV1 transfectants (RV-con, RV-KL, RV-KM, and RV-KH). After incubation at 37°C for 24 h, the CM-treated DU145 cells were used for morphological observation, cell lysate preparation, and total RNA extraction. For morphological studies, the cells were rinsed in warm PBS, fixed in methanol for 30 min, and stained with 0.1% (w/v) crystal violet. The stained cells were air-dried, viewed, and photographed under a phase-contrast microscope. For cell lysate preparation, the cells were washed twice with ice-cold PBS and lysed in 200 μl of RIPA buffer per well for 30 min at 4°C. The lysate was collected by cell scraping and centrifugation at 14,000 × g for 30 min at 4°C. The supernatant (cell lysate) was collected, and the total protein concentration was determined by the Bradford method.
Results
Establishment of hK7-expressing 22RV1 and DU145 prostate carcinoma cells. To overexpress hK7 in prostate cancer cell lines, we stably transfected the coding sequence of KLK7 into 22RV1 and DU145 cells. DU-K was from a pooled transfectants of DU145 clone, and three transfectants were selected from 22RV1 clone and were designated as RV-KL, RV-KM, and RV-KH based on their low, median, and high KLK7 transcription levels, respectively (see the Materials and Methods section). As hK7-negative controls, two transfectants were also selected from 22RV1 and DU145 cells transfected with empty vector and were named as RV-con and DU-con, respectively (see the Materials and Methods). RT-PCR analysis confirmed that pcDNA3.1-KLK7 transfectants (RV-KL, RV-KM, RV-KH, and DU-K) were all positive for KLK7 mRNA, whereas the RV-con and DU-con controls were negative (data not shown). As a 28-kDa secreted protease, overexpressed hK7 should be secreted into the cell culture medium. Western blot analysis with anti-hK7 polyclonal antibody revealed a specific 28-kDa band with different intensities in CMs from RV-KL, RV-KM, RV-KH, and DU-K cells, whereas no specific band was detected in CM from RV-con and DU-con cultures (Figure 1).

Effect of hK7 on cell proliferation. An MTT-based assay was used to determine proliferation and cell viability of the 22RV1 clones (A) and DU145 transfectants (B). Cells were seeded in 96-well culture plates as described in the Materials and Methods. Proliferative activities were measured by the MTT method, every 24 h for up to 96 h. The mean optical density at 570 nm for each cell type was plotted against time. Data shown are the mean±SD of triplicate cultures. The results were analyzed by one-way ANOVA, p>0.05.

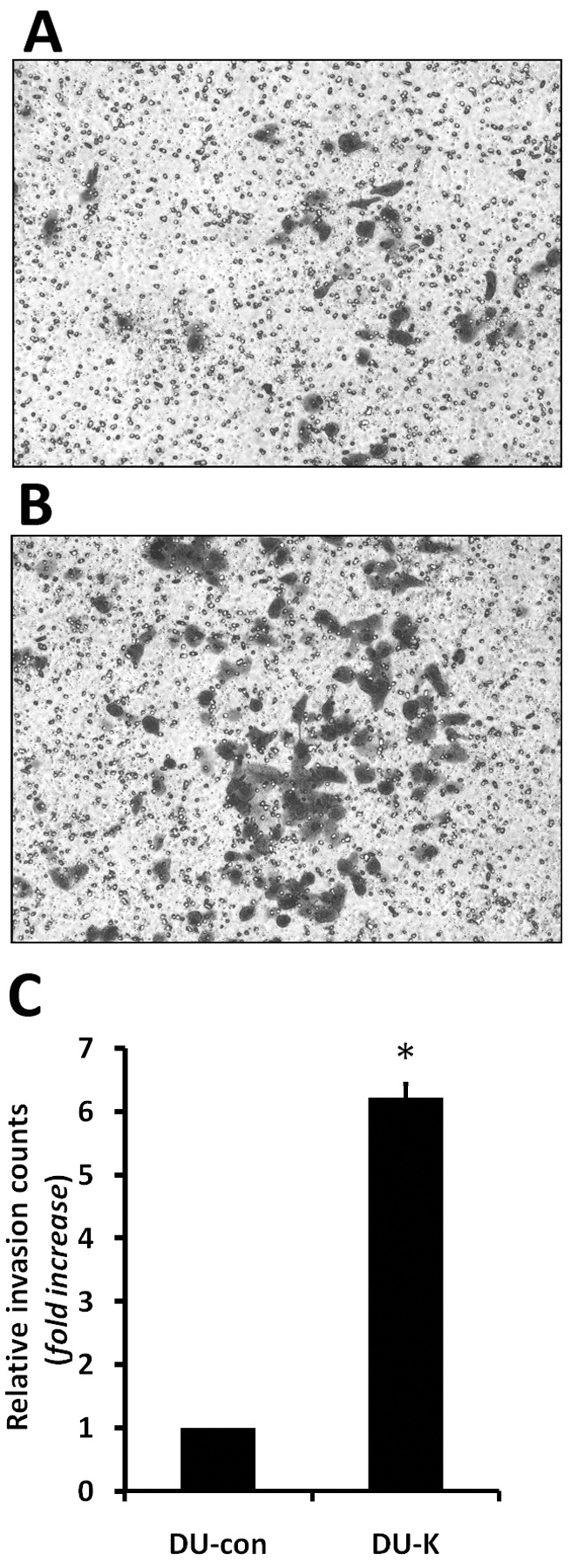
Effect of hK7 on the migration and invasiveness of DU145 cells in vitro. Following a 36-h incubation in Matrigel-coated Transwells, the cells that had invaded the Matrigel and migrated to the lower surface of the filter were stained with 0.1% crystal violet. A, B: Representative images of invading cells. The numbers of cells in predetermined fields were counted under a microscope, and the relative invasion was defined as the fold increase of DU-K cell number over the DU-con cell number. C: The bar represents the standard deviation of the mean of three independent experiments. The asterisk indicates a significant increase in the invasive ability of hK7-expressing DU-K cells compared with the control DU-con cells (one-way ANOVA, p<0.05).
Expression of hK7 did not alter proliferation of 22RV1 and DU145 cells. The growth rates of DU145 and 22RV1 transfectants were evaluated using an MTT-based proliferation assay. In three independent assays, the proliferative activity of RV-KL, RV-KM, RV-KH, and DU-K cells was similar to that of 22RV1 and DU145 cells transfected with vector alone (controls), respectively (Figure 2A and B, one-way ANOVA, p>0.05), suggesting that hK7 does not affect cell growth and proliferation in our cell system.
Expression of hK7 promoted invasiveness of DU145 cells in vitro. To assess the effect of hK7 on the invasiveness of prostate cancer cells, a Matrigel invasion assay was performed using DU145 cells, a moderately invasive prostate cancer cell line. In three separate experiments, the invasiveness of hK7-expressing DU-K cells was about 6-fold that of the control DU-con cells (Figure 3; one-way ANOVA, p<0.01). Thus, hK7 enhanced the migration and invasiveness of DU145 cells in vitro.
Expression of hK7 induced morphological changes in DU145 cells. In colony formation assay, following incubation in selective medium for 2 weeks, both DU-K and DU-con grew into visible cell colonies (Figure 4A). The general morphology of the DU-K colonies (Figure 4A-1) was quite distinct from that of the DU-con colonies (Figure 4A-2). The DU-K colonies (Figure 4A-2 and A-4) exhibited a relatively large and ‘loose’ appearance, whereas the DU-con colonies (Figure 4A-1 and -3) were generally small and ‘tight’. The morphologies of the DU-K and DU-con colonies were examined microscopically by determining the percentage of typical epithelia-like formation of cells tightly adhered to each other, which can be easily identified under a microscope (arrow in Figure 4A-3). The percentage of typical epithelia-like formation was significant lower in DU-K colonies compared with DU-con colonies (Figure 4B, p<0.01). Thus, hK7 appeared to have induced morphological changes in the DU145 cells.
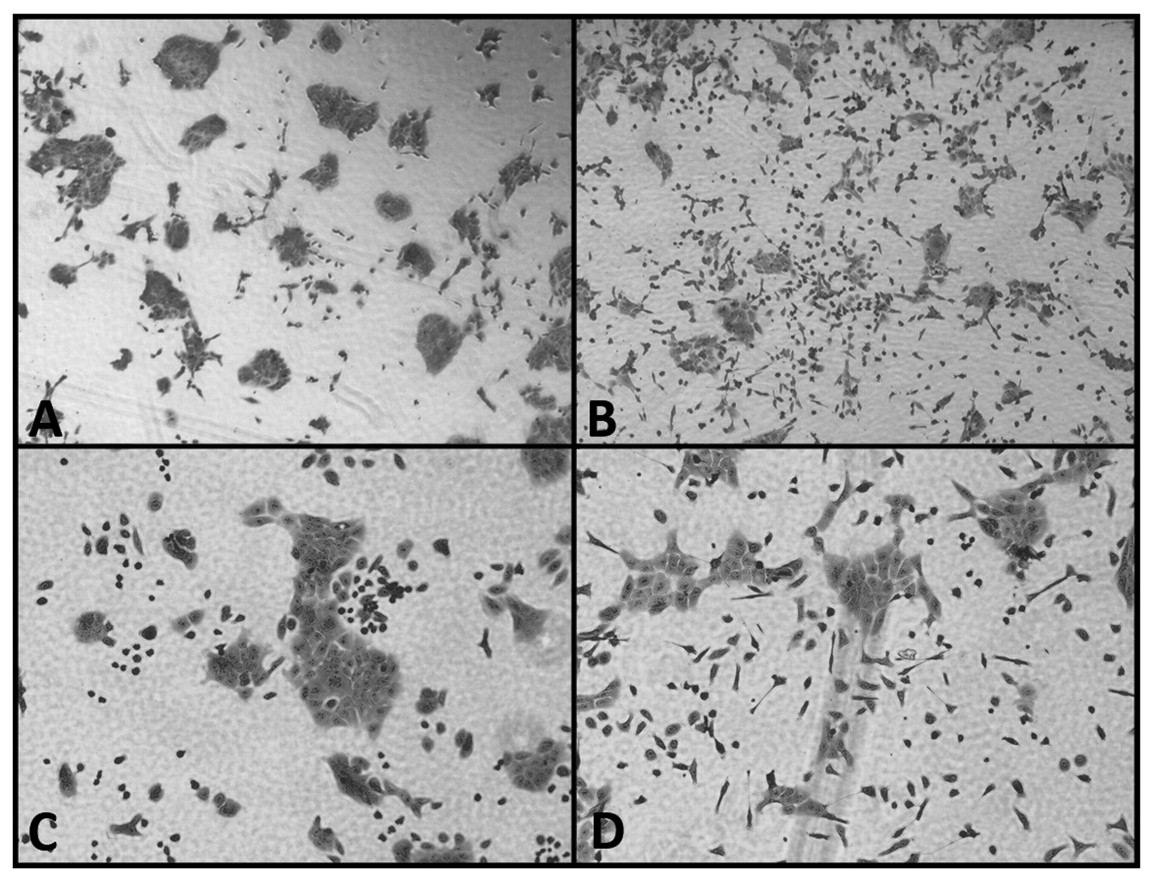
Conditioned medium containing hK7 induced morphological changes in DU145 cells. To further investigate the effect of hK7 on DU145 cell morphology, we treated DU145 cells with RV-KH CM containing a high concentration of hK7 and with RV-con CM lacking hK7, which served as the control. After 12 h, DU145 cells treated with the RV-KH CM became dissociated with spindle/elongated shapes and assumed a mesenchymal morphology (Figure 5B and D). These changes were not observed in DU145 cells treated by the hK7-negative CM of RV-con (Figure 5A and C).
Conditioned medium containing hK7 increased vimentin expression in DU145 cells. To further reveal the hK7-induced mesenchymal features in DU145 cells, we examined the expression of vimentin, an important mesenchymal marker, in DU145 cells treated with CMs containing different concentrations of hK7. Wild-type DU145 cells treated with CMs from RV-con, RV-KL, RV-KM, and RV-KH cultures, which contained different hK7 concentrations, were designated as RV-con/DU, RV-KL/DU, RV-KM/DU, and RV-KH/DU, respectively. The level of vimentin mRNA in the CM-treated DU145 cells was determined by quantitative real-time PCR and was normalized to the level of the housekeeping gene HPRT. The vimentin mRNA level in the CM-treated DU145 cells was positively correlated with the hK7 concentration in the CM. (Figure 6A). Furthermore, the levels of vimentin protein expression in cell lysates of RV-KH/DU and RV-con/DU cells were determined by Western blot analysis, revealing that the vimentin protein level in RV-KH CM-treated DU145 cells was significantly higher than that in RV-con CM-treated DU145 cells (Figure 6B). Thus, hK7 resulted in increased expression of vimentin in prostate cancer DU145 cells.
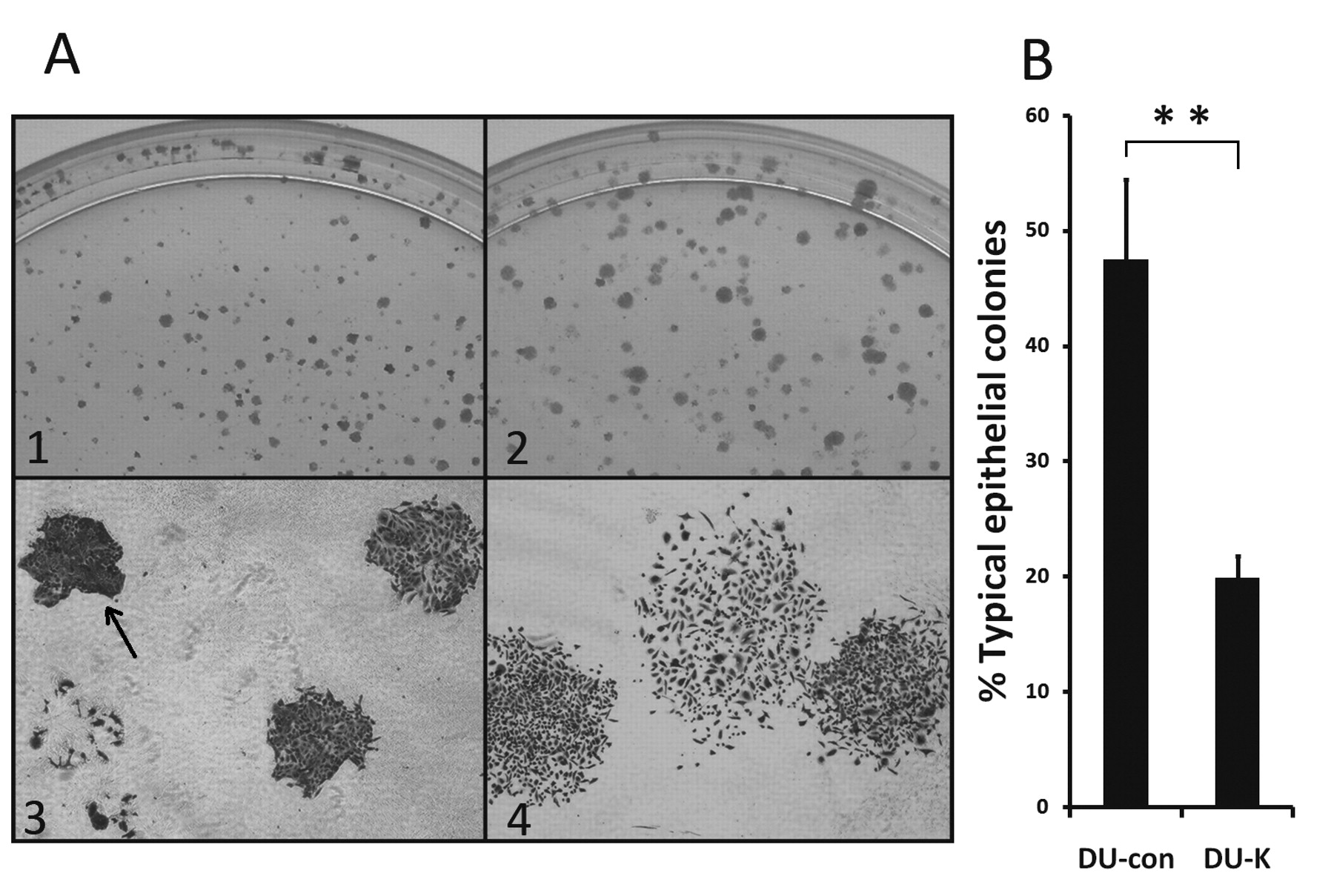
Morphological differences between the hK7-expressing DU-K colonies and the control DU-con colonies. A: A colony formation assay was performed as described in the Materials and Methods. The DU-con colonies (A-1) and DU-K colonies (A-2) showed distinctly different general morphology. Representative colonies of DU-con (A-3) and DU-K (A-4) were photographed under a microscope (original magnification, ×40). B: The percentages of typical epithelia-like colonies in the DU-con and DU-K colonies. Under phase-contrast microscopy, tightly formed colonies of polygonal, epithelia-like cells were denoted as epithelia-like colonies; these had typical cobblestone morphology and were easy to identify. Typical epithelia-like colonies (arrow in A-3) were counted in predetermined fields of the DU-con and DU-K colonies. Data are presented as mean percentages±SD of triplicate determinations. The percentages were significantly different between the DU-con and DU-K colonies (Student's t-test, **p<0.01).

Effect of hK7-containing conditioned medium (CM) on the morphology of DU145 cells. Parental DU145 tumor cells were treated with hK7-negative CM (A, C) or with CM containing abundant hK7 (B, D) for 12 h, as described in the Materials and Methods. The cells were stained with crystal violet and photographed under a phase-contrast microscope. Original magnification: ×40 in A and B, and ×200 in C and D.
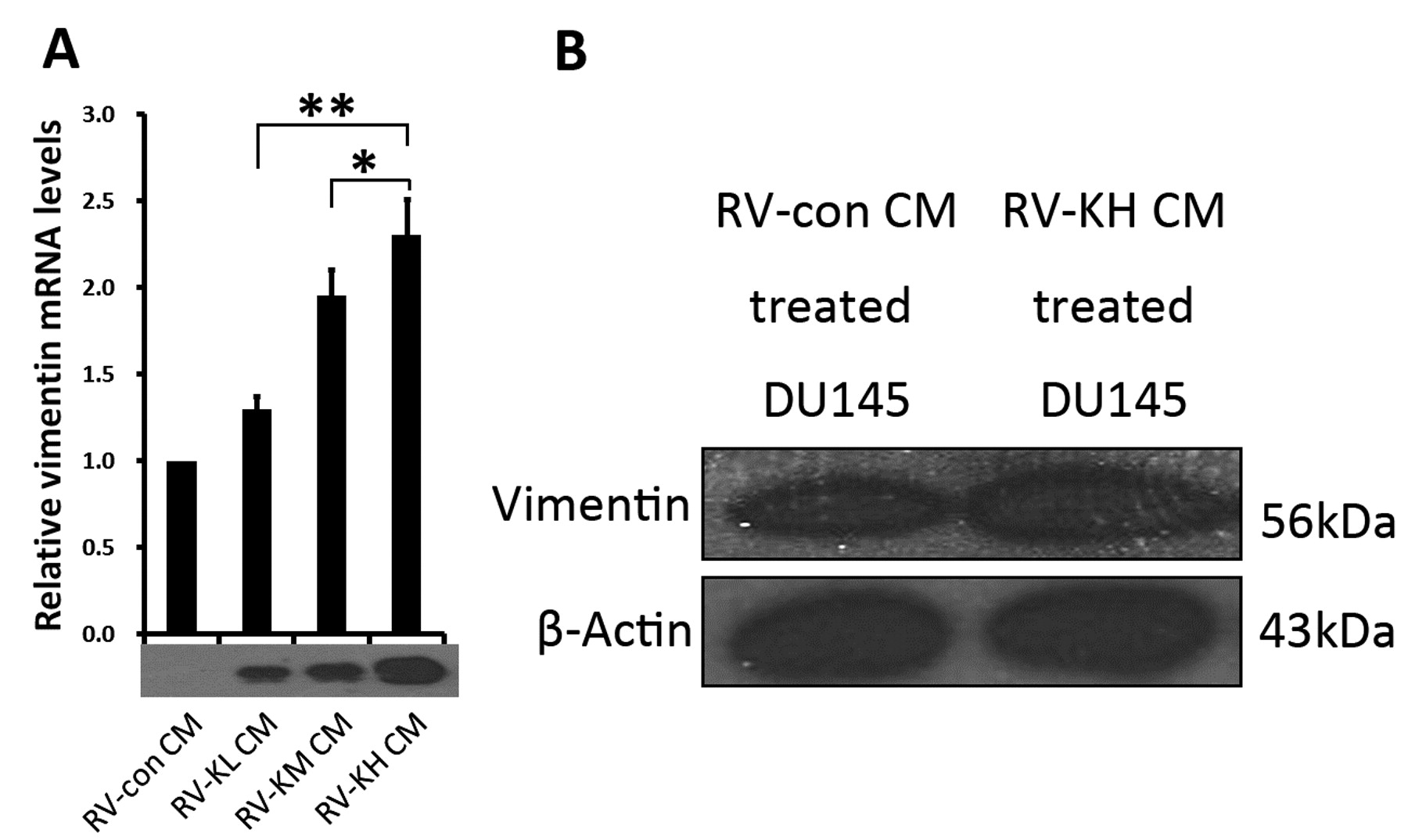
Conditioned medium (CM) containing hK7 induced vimentin expression in DU145 cells. A: The levels of vimentin mRNA expression in DU145 cells treated with CMs containing different concentrations of hK7 were quantified by real-time PCR and were normalized to the level of the housekeeping gene HPRT. The relative vimentin mRNA levels in DU145 cells treated with hK7-containing CMs (RV-KL CM, RV-KM CM, and RV-KH CM, respectively) are presented as fold increases compared with the level in DU145 cells treated with hK7-negative CM (RV-con CM) (*p<0.05; **p<0.01). B: Western blot analysis of vimentin protein expression in DU145 cells treated with hK7-negative CM (RV-con CM) and hK7-containing CM (RV-KH CM). The upper panel shows the specific 56-kDa vimentin band in DU145 cells treated with RV-KH CM. The lower panel shows the expression of β-actin as a loading control.
Discussion
Human K7, a secreted protease with chymotrypsin-like activity, has received attention recently because of its potential roles in cancer progression. Studies have suggested that hK7 may be related to a poor prognosis and progression of ovarian, cervical, breast, pancreatic, and intracranial malignancies. In the present study, we showed that hK7 promoted Matrigel invasion by DU145 prostate cancer cells in vitro. Moreover, hK7 led to mesenchymal morphological changes in DU145 cells, accompanied by the up-regulation of vimentin expression, which suggested an involvement of hK7 in the epithelial mesechymal transition (EMT) process. This evidence indicates that hK7 plays an important role in prostate cancer invasion and progression.
Proliferation assays in the present study revealed that hK7 exerted little effect on the growth of 22RV1 and DU145 cells; however, accumulating evidence indicates that hKs influence cancer cell proliferation through diverse mechanisms. Both hK2 and hK3 (prostate-specific antigen, PSA) have been reported to regulate cell growth by modulating the bioavailability and activity of factors in the insulin-like growth factor-insulin-like growth factor binding protein (IGF-IGFBP) axis (7), and hK2 and hK4 have been implicated in the activation of urokinase plasminogen activator (uPA)/uPA receptor (uPAR) systems (8), leading eventually to pericellular and extracellular matrix degradation and the release and/or activation of tumor-associated growth factors in vivo. In addition, some hKs may themselves serve as growth factors through the activation of protease-activated receptor signaling (9). In light of these previous reports, we cannot rule out the possibility that hK7 may affect cell proliferation in a tumor microenvironment, and this deserves further investigation in vivo.
Using co-transfection with plasmids expressing hK4, hK5, hK6, and hK7, Prezas and colleagues revealed that hK7 increased the invasiveness of OV-MZ-6 ovarian cancer cells in a cooperative manner in vivo (10). Another group demonstrated that hK7 overexpression in cultivated brain tumor cells significantly enhanced their invasive potential in a Matrigel invasion assay (11). In the present study, hK7 significantly increased DU145 prostate cancer cell invasion of Matrigel, in agreement with these previous reports. Although the effect of hK7 on malignant cells has been described, the mechanism by which hK7 increases cell invasiveness remains unclear. A recent report has provided a clue to the mechanism of hK7 action. Johnson and co-workers determined that hK7 can cleave the extracellular domain of E-cadherin to produce soluble fragments of E-cadherin; both the cleavage of E-cadherin and the increasing amount of E-cadherin fragments in the culture medium significantly decreased cell aggregation and increased cell invasiveness of pancreatic cancer cells in vitro (12). However, based on the results reported here, the shedding of E-cadherin may not completely explain the enhancement of invasiveness induced by hK7.
In addition to promoting cell invasiveness, hK7 also changed the morphology of DU145 cells in the present study. In the process of screening neo-resistant transfectants, we were surprised to note general morphological differences between DU-K and DU-con colonies. Microscopically, the DU-K colonies consisted of dissociated cells with mesenchymal-like shapes, whereas the DU-con colonies were composed mainly of cobblestone-shaped cells with tight cell−cell adhesion, suggesting that hK7 induced mesenchymal morphological changes in DU145 cells. Indeed, wild-type DU145 cells grown in CM that contained abundant hK7 became scattered and took on a spindle/elongated shape. Chunthapong et al. have demonstrated that spindle/elongated, fibroblastic-like DU145 cells are more invasive than round, epithelia-like cells (13), which agrees well with our results that hK7 induced mesenchymal morphological changes in DU145 cells and increased cell invasiveness.
Considering the mesenchymal morphological changes induced by hK7, we investigated hK7-induced changes in the expression of the mesenchymal intermediate filament vimentin, an important mesenchymal marker, and demonstrated that hK7 increased the expression of vimentin at both the mRNA and protein levels in DU145 cells. Given that DU145 cells express E-cadherin (13) and that hK7 can cleave the extracellular domain of E-cadherin (12), the up-regulation of vimentin expression may be explained as follows. Initially, hK7 would degrade the extracellular part of E-cadherin in DU145 cells, leading to cell−cell detachment and the destruction of E-cadherin−β-catenin complexes. The accumulated cytoplasmic β-catenin would enter the nucleus, where it would bind to T-cell factor/lymphoid enhancer factor 1 and up-regulate vimentin expression via the β-catenin/T-cell factor pathway (14). We plan further investigations to examine this proposed mechanism.
In the current study, hK7 led to to increased cell invasiveness, scattered cell growth, spindle/elongated cell shape, and up-regulated vimentin expression in DU145 cells. These changes are expected to be interrelated and correlated: hK7 would release the extracellular domain of E-cadherin, leading to vimentin up-regulation via the β-catenin/T-cell factor pathway, and vimentin would subsequently contribute to mesenchymal morphological changes in DU145 cells. In addition, the hK7-induced shedding of E-cadherin on the DU145 cell surface would reduce cell−cell adherence and result in dissociated cell growth. The hK7-induced increase in invasiveness of malignant cells, as well as the hK7-induced increase in Matrigel invasiveness of DU145 cells in the present study, may be correlated with one or more of the above steps. Moreover, the combination of the above changes strongly suggests that hK7 induces EMT-like changes in DU145 cells.
Theoretically, certain hKs may induce EMT, because some EMT-related factors are kallikrein substrates. For example, transforming growth factor-β (TGF-β) can induce EMT, and its latent form can be activated by PSA (15, 16). Therefore, some authors have predicted that particular hKs (e.g. hK3/PSA) may indirectly induce EMT by activating the latent TGF-β complex and thus promote the detachment and invasiveness of malignant cells. A recent study has shown that hK4 and PSA can induce EMT-like changes in PC-3 prostate cancer cells, with loss of E-cadherin expression and up-regulation of vimentin expression (17). In addition, Robin et al. have reported that the activation of uPAR can induce EMT in hypoxic breast cancer cells (18). As hK2 and hK4 are implicated in the activation of uPA/uPAR systems (8), we speculate that the hKs−uPAR−EMT axis may be a signaling pathway for correlating hK4 with EMT in PC-3 cells. The role of hKs in EMT is likely to be complicated and deserves further exploration.
In this study, we established hK7-expressing DU145 and 22RV1 cells. The hK7-expressing DU145 cells exhibited increased invasiveness and morphological changes. In contrast, the hK7-expressing 22RV1 cells showed no obvious phenotypic changes. According to a previous report (19), 22RV1 cells are not ideal for invasion assays owing to their extremely low inherent invasiveness, which is consistent with our results. We speculate that hK7 may affect only certain malignant cell types having specific signal pathways or existing in a specific differentiation state. In the present study, 22RV1 cells were used to generate cell clones with diverse hK7 expression levels in order to produce CM containing different concentrations of hK7. The hK7-induced EMT-like changes, along with increased invasiveness and vimentin up-regulation, occurred only in DU145 cells. Our findings provide new insight for understanding the involvement of hK7 in the poor prognosis and increased invasiveness of malignancies.
In conclusion, the present study demonstrated that hK7 promotes cancer cell migration and invasiveness and induces EMT-like changes in DU145 prostate tumor cells in vitro, suggesting that hK7 accelerates prostate cancer progression through the induction of EMT. As prostate carcinoma metastasis remains a major cause of failure for the clinical treatment and management of this disease and as hK7 appears to be important in mediating the invasion and metastatic spread of prostate cancer, our findings suggest an attractive approach for the development of novel therapeutic treatments for this highly metastatic disease. Thus, hK7-targeted therapy merits further investigation as a promising clinical intervention for prostate cancer that cannot be cured by conventional therapies.
Acknowledgements
This project was carried out at the Institute of Urology, First University Hospital of Guangxi Medical University and the Institute for Molecular Biology, Nankai University. This work was supported by grants from the National Natural Science Foundation of China (No. 30260110/C03030305) and the First University Hospital of Guangxi Medical University.
- Received January 3, 2010.
- Revision received June 21, 2010.
- Accepted June 25, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.