


Abstract
ErbB receptors are crucial for development and evolution and have been intensely pursued as targets for cancer therapeutics. Inhibiting the signaling activity of individual receptors in this family has advanced human cancer treatment. However, actual curative effects of the existing anti-ErbB therapeutics are still insufficient. A large percentage of patients who are initially responsive to ErbB receptor-targeted therapies later become resistant. Mechanisms responsible for tumor resistance to ErbB-targeted agents are as follows: many epidermal growth factor receptor (EGFR)- and HER2-targeted therapies cannot inhibit signaling through the ErbB receptor heterodimer, and anti-EGFR agents can suppress extracellular signal-related kinase (ERK) signal proliferation but not protein kinase B/Akt survival signals. ErbB ligand-based targeted therapy against HB-EGF or amphiregulin may overcome such obstacles. Here we discuss the efficacy of CRM197, a specific inhibitor of HB-EGF, and its possible clinical adaptation in combination with conventional chemotherapeutic agents in cancer therapy.
Conventional cancer therapy is predominantly based on cytotoxic chemotherapeutic agents. Cytotoxic events occur due to disruption of various aspects of DNA synthesis and repair or due to the disturbance of mitosis. As these cellular processes are common to all dividing cells, most chemotherapeutic agents frequently cause substantial adverse side-effects.
Selective inhibition of molecular targets that play pivotal roles in carcinogenesis, tumor maintenance, and metastasis offers the promise of maximum benefit with minimum side-effects. A premise of targeted therapy is that a cancer cell is dependent on a contracted signaling network that includes one or more hyperactive pathways. Kinases are key molecules in signal transduction, and the ErbB family of receptor tyrosine kinases (RTKs) is a prime example of targets in cancer therapy.
The ErbB family of RTKs (Figure 1) includes epidermal growth factor receptor (EGFR, also known as ErbB1/HER1), ErbB2/Neu/HER2, ErbB3/HER3, and ErbB4/HER4, and these mediate major cellular functions, such as proliferation, differentiation, motility, and survival (1). They have been implicated in the development and progression of most common human epithelial malignancies (2). Thus, ErbB receptors have been intensely studied as therapeutic targets.
In this review, we have outlined our understanding of ErbB receptors and their ligands, especially mechanisms responsible for tumor resistance to anti-ErbB therapies. We discuss alternative strategies, including ErbB ligand-based targeted therapy, which might be more effective in cancer treatment, and also discuss clinical adaptations in combination with conventional chemotherapeutic agents.
The ErbB Receptors and their Ligands
The ErbB family of RTKs consists of transmembrane proteins that share high homology in the extracellular ligand-binding domain, short hydrophobic transmembrane region, and cytoplasmic tyrosine kinase domain (3). Receptor dimerization is essential for activation of the ErbB signaling network (3, 4). However, ErbB receptors normally exist as inactive monomers with the molecules folded to prevent dimerization (5, 6). A family of ligands, EGF-related peptide growth factors, binds the extracellular domain of ErbB receptors to initiate a conformational rearrangement, exposing the dimerization domain that leads to formation of both homodimers (between two molecules of the same ErbB receptor) and heterodimers (between two different ErbB receptors) (7). Dimerization consequently stimulates the intrinsic tyrosine kinase activity of the receptors and triggers autophosphorylation of specific tyrosine residues within the cytoplasmic domain. Phosphorylation allows the recruitment and activation of downstream proteins, and a signaling cascade is initiated (3, 8). Several different homodimer and heterodimer pairings are possible between the four receptors (Figure 1), allowing a diverse range of signaling molecules and pathways to be activated (8). However, ErbB3 is not kinase active, cannot form homodimers, and thus only forms heterodimers (9). To date, no ligand has been identified for ErbB2/HER2 (6, 10). ErbB2/HER2 and ErbB3 function as preferred heterodimer partners of other ErbB receptors. Dimerization subsequently up-regulates downstream signaling cascades, including mitogen-activated protein kinase (MAPK), phosphoinositol 3-kinase/v-akt murine thymoma viral oncogene homolog (PI3K/Akt), and mammalian target of rapamycin pathways (8, 11).
These receptors receive extracellular signals from small peptide ligands, which include the following: (i) EGF, transforming growth factor (TGF)-α, amphiregulin (AR), and epigen, which bind specifically to EGFR; (ii) heparin-binding EGF-like growth factor (HB-EGF), epiregulin, and betacellulin, which exhibit dual specificity by binding both EGFR and ErbB4; and (iii) neuregulins, which include NRG1 and NRG2 that bind both ErbB3 and ErbB4, as well as NRG3 and NRG4 that bind only ErbB4 (12) (Figure 1).
Response and Resistance to ErbB Receptor-targeted Therapy
EGFR and other ErbB receptors have been especially recognized as target molecules for cancer therapy because activation or mutation of these molecules correlates strongly with the pathogenesis and poor prognosis of many types of human cancer. Various tyrosine kinase inhibitors targeting EGFR (e.g. erlotinib: Tarceva), as well as monoclonal antibodies targeting EGFR (e.g. cetuximab: Erbitux) and ErbB2/HER2 (e.g. trastuzumab: Herceptin) have been developed and have progressed to clinical applications; however, clinical studies using EGFR antagonists have not always resulted in favorable clinical outcomes (13). A large percentage of patients who are initially responsive to ErbB receptor-targeted therapies experience tumor recurrence and become refractory to therapy (14). One of the reasons for resistance is that EGFR and ErbB2/HER2 form complexes with ErbB3 and other signal receptors. Proliferation of cancer cells is subsequently accelerated by these complexes, whose formation cannot be inhibited by targeted therapies against EGFR and ErbB2/HER2. Another reason is that anti-EGFR drugs can reduce proliferation of extracellular signal-related kinase (ERK) signals located downstream of EGFR, but they cannot suppress protein kinase B/Akt survival signals. Resistance may arise in tumor cells through allelic and adaptive changes, leading to PI3K activation through other receptor tyrosine kinases. Down-regulation of insulin-like growth factor binding proteins 3 and 4, which are negative regulators of IGF-I receptor (IGF-IR) signaling, causes activation of IGF-IR and the PI3K/Akt pathway and contributes to the development of resistance to EGFR inhibitors (15).
These observations may provide guidelines for developing more efficient therapeutic approaches. To improve resistance, multitargeted EGFR inhibitors and combined inhibition of EGFR and Akt are proposed (16).
ErbB Ligand-based Targeted Therapy
We have proposed EGFR ligand-based targeted therapy for cancer. The belief that ligand targeting is less effective than receptor targeting in the EGF signaling network has delayed the development of such medicines, but our research has revealed that this notion is not true. The aberrant enhancement of EGFR ligand expression is speculated to be one of various molecular mechanisms accounting for acquired resistance to EGFR antagonists. We have reported evidence indicating that EGFR ligands deserve considerable attention as potential targets for cancer therapy, and discussed EGFR signaling inhibition strategies directed at EGFR ligands, such as HB-EGF and AR (17). HB-EGF expression is dominantly elevated in ovarian, gastric, and breast cancer, as well as in melanoma and glioblastoma. AR expression is primarily enhanced in pancreatic, colon, and prostate cancer, as well as in renal cell carcinoma and cholangiocarcinoma (18). In cell lines with dominant HB-EGF expression, transfection of small interfering RNAs (siRNAs) for HB-EGF increases the number of apoptotic cells and suppresses activation of EGFR and ERK, whereas transfection of siRNAs for other EGFR ligands shows no effects. Similarly, apoptosis and attenuation of EGFR and ERK signals in cell lines with abundant AR expression are significantly mediated by AR inhibition.
Receptor dimerization is essential for activation of the ErbB signaling network. When one receptor is functionally inactivated, its function can be substituted by another ErbB receptor. MDA-MB-468 breast cancer cells secrete abundant amounts of the soluble form of HB-EGF (sHB-EGF) and form EGFR/HER2 complexes; this complex formation is enhanced by trastuzumab but reduced by CRM197, a specific HB-EGF inhibitor. CRM197 attenuates phosphorylation of ERK and Akt and leads to significant apoptosis compared with trastuzumab. We supposed that ligand-induced ErbB receptor dimerization plays pivotal roles in retrieving the intracellular signal for cell survival against therapy targeting EGFR or HER2 alone, and that targeting of a dominantly expressed EGFR ligand is an available strategy for cancer therapy.
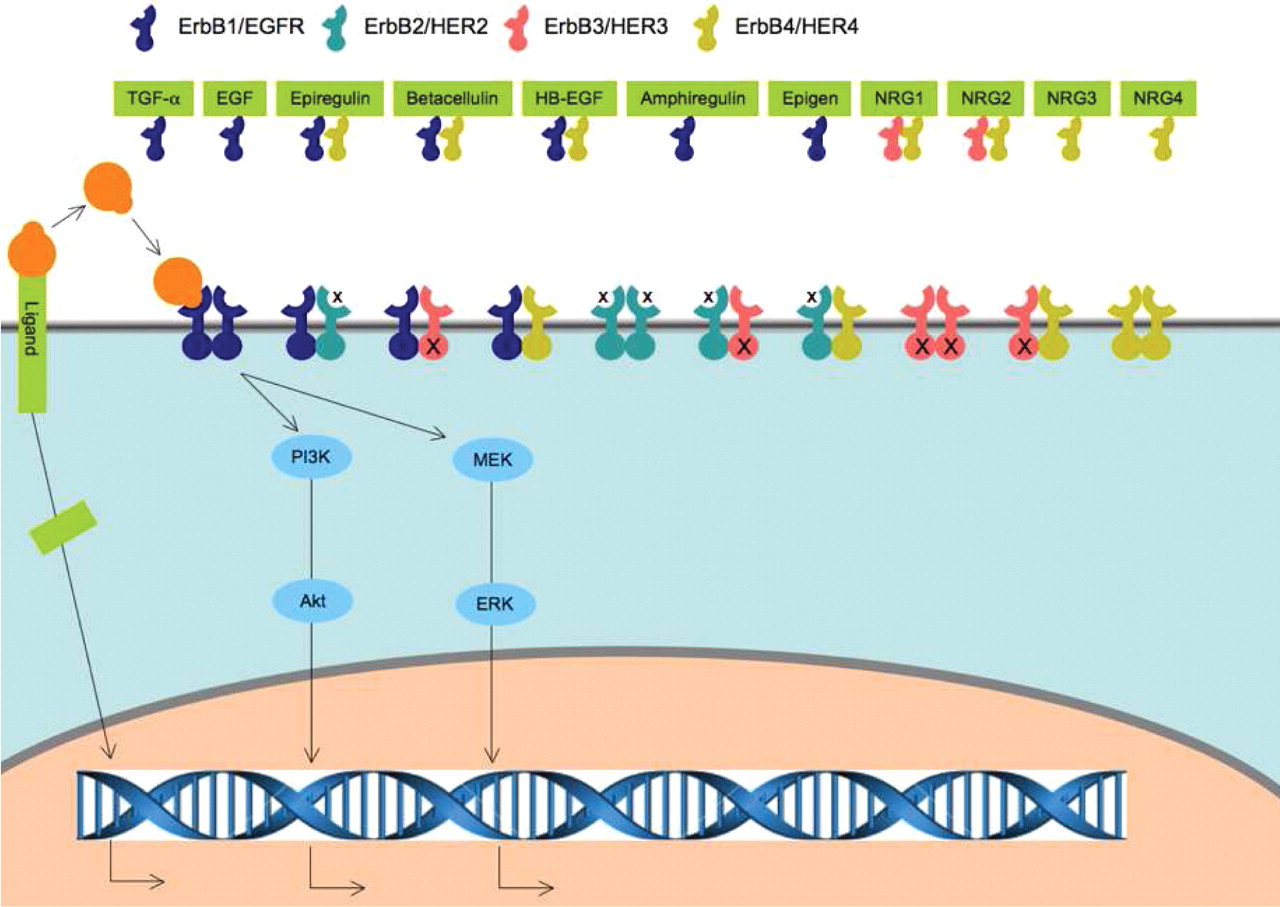
ErbB receptors and their ligands. The ErbB family includes four receptors, namely EGFR/ErbB1/HER1, ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4. EGFR, HER2, and ErbB4 have a cytoplasmic tyrosine kinase domain that is activated by dimerization. The ErbB3 kinase domain differs due to the lack of tyrosine kinase function. A total of eleven ligands have been identified. These include EGF, transforming growth factor (TGF)-α, amphiregulin (AR), and epigen (EPGN), which bind specifically to EGFR;, heparin-binding EGF-like growth factor (HB-EGF), epiregulin (EPR), and betacellulin (BTC), which exhibit dual specificity by binding both EGFR and ErbB4; and neuregulins, which include NRG1 and NRG2 that bind both ErbB3 and ErbB4, as well as NRG3 and NRG4 that bind only ErbB4. No ligand has been identified for HER2. The membrane-anchored ligand is cleaved at the cell surface by a protease to yield the soluble form of HB-EGF (sHB-EGF) via a mechanism known as ectodomain shedding. The carboxyl-terminal fragment (CTF) of the ligand translocates from the plasma membrane to the nucleus and regulates the cell cycle. ErbB family receptors receive extracellular signals from small peptide ligands and subsequently up-regulate downstream signaling cascades, including MAPK and PI3K/Akt pathways.
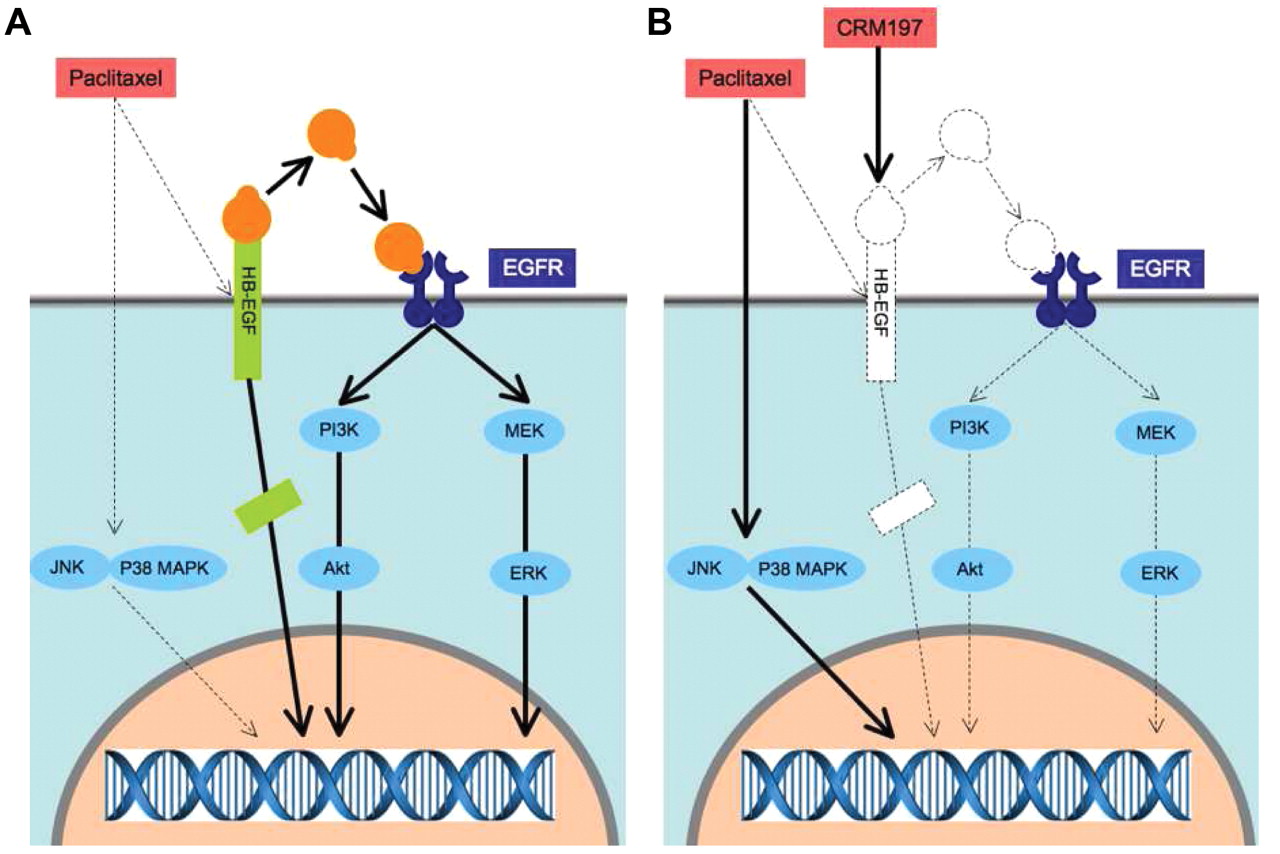
The mechanism involved in the synergistic antitumor effect of paclitaxel in combination with CRM197. A: Paclitaxel evokes proapoptotic signals, including JNK and p38 MAPK. Paclitaxel also promotes ectodomain shedding of proHB-EGF, resulting in activation of ERK and Akt as antiapoptotic signals through EGFR transactivation. HB-EGF-CTF translocates to the nucleus to transmit cell growth signals. The enhancement of HB-EGF expression possibly results in the acquisition of resistance to paclitaxel. B: CRM197 blocks EGFR transactivation, leading to suppression of ERK and Akt activation, and may inhibit the shift of HB-EGF-CTF to the nucleus. Therefore, in the presence of CRM197, paclitaxel activation of antiapoptotic signals through ectodomain shedding of proHB-EGF is reduced. The combination of paclitaxel with CRM197 induces synergistic antitumor effects in cancer therapy.
HB-EGF as a Targeted Molecule
HB-EGF directly binds and activates EGFR and indirectly transactivates HER2, ErbB3, and ErbB4 by forming a heterodimer with EGFR (19). HB-EGF is initially synthesized as a transmembrane protein similar to other members of the EGF family of growth factors (20). The membrane-anchored form of HB-EGF (proHB-EGF) is cleaved at the cell surface by a protease to yield sHB-EGF via a mechanism known as ectodomain shedding (21). Members of the ADAM (a disintegrin and metalloprotease) family of proteases, such as ADAM9, ADAM10, ADAM12, and ADAM17, or other metalloproteases, are involved in this process (22). Various signaling pathways are activated following ectodomain shedding of proHB-EGF (23, 24). sHB-EGF is a potent mitogen and chemoattractant for many different cell types. Ectodomain shedding is not only a key event of receptor cross-talk, but also a novel intracellular signaling by the HB-EGF carboxyl-terminal fragment (CTF). Recent studies have reported that HB-EGF-CTF translocates from the plasma membrane to the nucleus and regulates the cell cycle (25, 26). HB-EGF-CTF can associate with Bcl2-associated athanogene 1 (BAG-1), promyelocytic leukemia zinc finger (PLZF), and B-cell leukemia 6 (Bcl6). Interaction between BAG-1, a prosurvival co-chaperone, and HB-EGF-CTF leads to attenuation of cell adhesion, resistance to apoptosis, and enhancement of sHB-EGF expression (27). PLZF and Bcl6 are transcriptional repressors that function as negative regulators within the cell cycle.
Increasing evidence suggests a critical role for HB-EGF in tumor cell growth and progression. HB-EGF gene expression in cancerous tissue and HB-EGF protein levels have been reported to be significantly elevated in patients' ascitic fluid (28, 29). Tumor formation by human ovarian cancer cells is enhanced by exogenous expression of proHB-EGF and completely blocked by siRNA for HB-EGF and CRM197 (28). CRM197 is a nontoxic mutant of diphtheria toxin and shares immunological properties with the native molecule. CRM197 binds to human HB-EGF and blocks its mitogenic activity caused by inhibition of EGFR binding (30). In patients with ovarian cancer, high HB-EGF expression is significantly associated with poor clinical outcome (31). According to these studies, HB-EGF is a promising target for cancer cells with dominant HB-EGF expression. We investigated the antitumor effects of CRM197 on ovarian cancer cells by evaluating proliferation of human ovarian cancer cell lines, SKOV3, RMG1, and OVMG1, subcutaneously implanted into nude mice. CRM197 significantly suppresses peritoneal dissemination in the nude mice peritoneally injected with ovarian cancer cells (32). CRM197 has previously been used for patients with cancer in a clinical research study (33). With the approval of the Ethics Committee, a phase I study on the use of CRM197 is being conducted at the Fukuoka University for patients with advanced ovarian cancer.
Synergistic Antitumor Effect of CRM197 in Combination with Conventional Chemotherapeutic Agents
Paclitaxel is widely used as a therapeutic agent for various type of cancer, including ovarian, breast, and lung cancer. Paclitaxel promotes ectodomain shedding of proHB-EGF as well as induces transient ERK activation and sustained activation of Jun-terminal kinases (JNK) and p38 mitogen-activated protein kinase (MAPK) through EGFR transactivation in SKOV3 cells (Figure 2A). HB-EGF overexpression in paclitaxel-treated SKOV3 cells resulted in modulation of paclitaxel-evoked MAPK signaling, including marked activation of ERK and Akt and minimized activation of JNK and p38 MAPK, indicating that HB-EGF is involved in drug sensitivity through the balance of paclitaxel-induced antiapoptotic and proapoptotic signals. The combination of paclitaxel with CRM197 had an inhibitory effect on cell proliferation and enhances apoptosis via inhibition of ERK and Akt activation and stimulation of p38 MAPK and JNK activation. More prominently, administration of paclitaxel in combination with CRM197 resulted in synergistic antitumor effects in SKOV3 cells and in SKOV3 cells overexpressing HB-EGF in xenografted mice (34) (Figure 2 B).
Although paclitaxel promotes ectodomain shedding of proHB-EGF, resulting in the synergistic antitumor effect of CRM197, the effect is not apparent with all chemotherapeutic agents because the degree of ectodomain shedding of proHB-EGF is different with different chemotherapeutic agents (unpublished data). On the other hand, reduction of HB-EGF expression attenuated expression of matrix metalloprotease-2 and vascular endothelial growth factor (32). The most suitable partner in a combination therapy with CRM197 remains to be determined.
Future Aspects
A novel strategy targeting multiple ligands of the ErbB family of receptors using a bispecific ligand trap, RB200, has been reported (35, 36). RB200 was designed as a chimeric molecule composed of full-length extracellular domains of EGFR and HER3 fused with the Fc domain of human immunoglobulin G1. RB200 bound EGFR (EGF, TGF-α, and HB-EGF) as well as HER3 ligands (NRG1). RB200 exhibited inhibitory effects on cell growth in nine tumor cell lines and in vivo antitumor efficacy in two xenograft models. EGFR and HER3 ligand traps may be potent tools for cancer therapy.
AR is also a potential target candidate for an ErbB ligand-based targeted therapy. In future, we intend to focus on the development of an AR-specific inhibitor, as well as another HB-EGF inhibitor using chemical compound screening.
We have already started a phase I clinical trial of CRM197 administration for advanced or recurrent ovarian cancer, and are designing a study on a novel combination therapy including CRM197 and another chemotherapeutic agent.
Ongoing efforts to understand how the ErbB system is involved in carcinogenesis may lead to new approaches for cancer therapy. These findings will help improve clinical outcomes in cancer patients.
- Received June 8, 2010.
- Revision received June 26, 2010.
- Accepted July 2, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A Possible Clinical Adaptation of CRM197 in Combination with Conventional Chemotherapeutic Agents for Ovarian Cancer
- Suppression of Amphiregulin/Epidermal Growth Factor Receptor Signals Contributes to the Protective Effects of Quercetin in Cirrhotic Rats
- Assessment of HB-EGF Levels in Peritoneal Fluid and Serum of Ovarian Cancer Patients using ELISA