


Abstract
Background: Promoter hypermethylation and global hypomethylation in the human genome are hallmarks of most cancers. Detection of aberrant methylation in white blood cells (WBC) has been suggested as a marker for cancer development, but has not been extensively investigated. This study was carried out to determine whether aberrant methylation in WBC DNA can be used as a surrogate biomarker for breast cancer risk. Patients and Methods: Promoter hypermethylation of 8 tumor suppressor genes (RASSF1A, APC, HIN1, BRCA1, CYCLIND2, RARβ, CDH1 and TWIST1) and DNA methylation for three repetitive elements (LINE1, Sat2 and Alu) were analyzed in invasive ductal carcinoma of the breast, paired adjacent normal tissue and WBC from 40 breast cancer patients by the MethyLight assay. Methylation in WBC from 40 controls was also analyzed. Results: Tumor and adjacent tissues showed frequent hypermethylation for all genes tested, while WBC DNA was rarely hypermethylated. For HIN1, RASSF1A, APC and TWIST1, there was agreement between hypermethylation in tumor and adjacent tissues (p=0.04, p=0.02, p=0.005 and p<0.0001, respectively). DNA methylation for the three repetitive elements was lower in tumor compared to adjacent tissue and WBC DNA. Significant correlations in the methylation of Sat2M1 between tumor and adjacent tissues and WBC DNA were found (p<0.0001 and p=0.046, respectively). There was also a significant difference in methylation of Sat2M1 between cases and controls (p=0.01). Conclusion: These results suggest that further studies of WBC methylation, including prospective studies, may provide biomarkers of breast cancer risk.
- Breast cancer
- promoter hypermethylation
- genomic methylation
- tumor suppressor genes
- repetitive elements
- WBC DNA
Breast cancer is the most common cancer and second cause of cancer-death among females in most Western countries where there is an overall lifetime risk of >10% of developing breast cancer (1). Although diagnosis by screening mammography is believed to be responsible for the significant decline in breast cancer mortality, the limitations of mammography are well recognized, especially for women with premenopausal breast cancer (2). Thus, alternative approaches to breast cancer detection are clearly needed for improving diagnosis/prognosis.
Epigenetic alterations, such as DNA methylation are widely accepted as a potential source of early biomarkers for better diagnosis/prognosis (3, 4). Many tumors exhibit excessive methylation of CpG islands that are found near the transcriptional promoters of mammalian genes (5). Indeed, hypermethylation of CpG islands in specific gene promoters is thought to contribute to carcinogenesis through transcriptional silencing of tumor suppressor gene expression, leading to the initiation and progression of cancer (6, 7).
Aberrant methylation is frequently found in breast tumors with more than 40 genes shown to be inactivated by promoter CpG island hypermethylation (8). Among these genes, RASSF1A (9, 10), CDH1 (11), RARβ (12, 13), CYCLIND2 (14, 15), TWIST1 (16), BRCA1 (17, 18), HIN1 (19) and APC (20, 21) are frequently methylated. Different studies have confirmed the hypothesis that aberrant methylation at specific genes contributes to the malignant phenotype of breast cancer. In addition, it has been increasingly recognized that tumor DNA can be found in the bloodstream of cancer patients and that this DNA frequently contains the same genetic and epigenetic aberrations as DNA isolated from an individual's tumor (3, 22-24). This suggests that detection of tumor DNA in the blood may serve as an early and more accessible marker of diagnosis and prognosis of breast cancer. However, the frequency of aberrant methylation in white blood cells (WBC) has not been extensively investigated.
Diffuse genomic hypomethylation is another hallmark of most cancers (25) including breast cancer (26, 27). This loss of DNA methylation may promote chromosomal instability (28) and increased cell proliferation through alterations in the expression of proto-oncogenes (29). Several investigators have used the DNA methylation of repetitive elements as a biomarker for global methylation status (30). Decreased genomic DNA methylation levels, assessed by PCR analysis of surrogate repetitive DNA elements including LINE1, Sat2M1 and Alu have been shown to be tightly associated with various types of cancer (31-33). Moreover, global hypomethylation in peripheral blood DNA was suggested as an independent risk factor for bladder, colorectal and head and neck cancers (34-36). These results suggest that WBC DNA may be a potential surrogate biomarker for systemic genome methylation status.
In the present study, we examined the methylation status of BRCA1, HIN1, RASSF1A, CDH1, RARβ, APC, TWIST1 and CyclinD2 and in LINE1, Sat2M1 and AluM2 in breast tumors, paired adjacent normal tissues and WBC using the MethyLight assay. The specific aims were to: (1) determine aberrant hyper- and hypo-methylation of selected genes/repetitive DNA elements in invasive ductal carcinoma of the breast, paired, adjacent normal tissue and WBC, (2) determine the correlation between methylation status in tumor and non-tumor tissues and (3) compare methylation levels in WBC DNA between cases and unaffected controls.
Patients and Methods
Study population and sample collection. Surgically resected specimens from breast carcinomas, matched adjacent normal tissues and peripheral blood samples were collected from 40 breast cancer patients undergoing mastectomy in the Oncology Institute, University of Istanbul in Turkey between 1991 and 1997 after approval of the study by the Institutional Human Ethics Committee. Frozen tissues and blood samples were obtained at the time of surgery from patients who had not been treated with chemotherapy prior to surgery. All patients were diagnosed with invasive ductal carcinoma with tumors >2 cm. All specimens were processed within 3-4 h of collection. The corresponding adjacent normal tissue sample was selected 12-15 cm away from the site at which the primary tumor was sampled. Forty ethnicity-matched healthy women, mostly employees of the Oncology Institute provided blood specimens during the same time interval as cases. These healthy women did not have evidence of disease at the time of enrollment in the study.
General and clinicopathological characteristics in breast cancer patients and controls.
DNA preparation/sodium bisulfite conversion. Ten ml whole blood and 0.25-0.5 cm3 fresh-frozen tissues were used for DNA isolation. A phenol/chloroform extraction procedure was used after overnight incubation with proteinase K at 37°C for DNA isolation. DNA bisulfite treatment was carried out using the EZ DNA Methylation-Gold Kit™ (Zymo Research, CA, USA) according to the manufacturer's instruction. Briefly, tissue and blood DNAs (1 μg) denatured at 98°C for 10 min were incubated with modification reagents for 2.5 h at 64°C and cleaned and desulphonated. The DNA was stored immediately at −20°C.
Methylation analysis. After sodium bisulfite conversion, genomic DNA was analyzed by the MethyLight technique as described previously (30, 37). In brief, DNA was PCR-amplified in a 7900HT Fast Real-Time PCR System (Applied Biosystems, CA, USA) using TaqMan® Universal PCR Master Mix (Applied Biosystems) under the following conditions; 95°C for 10 min, followed by 45 cycles at 95°C for 15 s and 60°C for 1.5 min. The primers and probes for BRCA1, HIN1, RASSF1A, CDH1, RARβ, APC, TWIST1, CYCLIND2, β-actin (ACTB) and repetitive elements including LINE1, Sat2, Alu and AluC4 were previously described (30, 37-39). ACTB and AluC4 were used to normalize for the amount of input DNA for the genespecific and global assays, respectively. Specificity of the reactions for methylated DNA was confirmed separately using CpGenome™
Universal methylated DNA (Chemicon, MA, USA) and unmethylated human sperm DNA. TaqMan PCR reactions with primers specific for the bisulfite-converted methylated sequence for a particular locus and with the ACTB reference primers were performed separately. The values obtained in these two TaqMan analyses were used as a measure of the degree of methylation at that locus. Relative quantification was determined based on the threshold cycles of the gene of interest and of the internal reference gene. The percentage of methylation at a specific locus was calculated by the 2-ΔΔCT method, where ΔΔCT= (CT,Target−CT,Reference)sample − (CT,Target−CT,Reference)fully methylatedDNA (40) and multiplied by 100. Samples containing ≥1% fully methylated molecules were designated as methylated, whereas samples containing <1% were designated as unmethylated. For the 2-ΔΔCT method to be valid, the amplification efficiencies of the test genes and reference gene, ACTB and the repetitive elements, LINE1, Sat2 or Alu and reference element AluC4 must be approximately equal. This was examined using real-time PCR and Taqman detection of serial dilutions of DNA with a 100-fold range and gene-specific primers of each gene/repetitive element and ACTB/AluC4. The ΔCT (CT,Target gene−CT,Reference) was calculated for each DNA dilution and a plot of the log DNA dilution vs. ΔCT was made. All amplification efficiencies were similar (data not shown). All samples were assayed in duplicate and the MethyLight assay was further validated by using mixtures of fully methylated and unmethylated DNA to give 0, 1, 5, 10, 25, 50 or 100% methylation. The range of intra-assay coefficients of variation (CVs) was 0.3-1.5.
Promoter hypermethylation in breast tumor, paired normal adjacent tissue and WBC DNAs from breast cancer cases.
Statistical analysis. Statistical analysis was performed using SAS 8.1 for Windows statistical package (SAS Institute, Cary, NC, USA). Gene-specific methylation status was categorized as positive if values were ≥1% but data were also evaluated by categories, <1%, ≥1 to <4%, ≥4 to <10%, and ≥10%. Fisher's exact tests were calculated to examine the agreement of gene-specific methylation status between DNAs from different sources (tumor, adjacent tissues and blood) among cases and the differences of promoter methylation status by clinical characteristics (menopausal status, histological stage, and metastasis status) among cases, and between cases and control status. ANOVA tests adjusted for age were used to test for the differences in methylation levels of LINE1, Sat2 or Alu, used as continuous variables, between cases and controls. Spearman correlation coefficient was used to determine the correlation of methylation levels of LINE1, Sat2 or Alu between different sources of DNA. All statistical tests were based on two-tailed probability.
Results
We performed the MethyLight analysis to evaluate the promoter hypermethylation status of 8 tumor suppressor genes and DNA methylation for three repetitive elements on 147 DNA samples from tumor tissue (n=40), adjacent tissue (n=27) and WBC (n=40 for both cases and controls).
Characteristics of the study population. The demographics of the study population are shown in Table I. The mean ages of patients and controls were 50.8±10.8 (range, 34-73) and 48.3±8.6 (range, 33-70) years, respectively. The mean overall age of the two groups was not significantly different (p=0.26). There were no significant differences in the distribution of age groups and menopausal status between cases and controls.
Promoter hypermethylation in tumor and adjacent tissues. The results of promoter hypermethylation levels for each sample are shown in Figure 1. Tumor and adjacent normal tissues were frequently hypermethylated for all the tumor suppressor genes tested. In particular, three genes (RASSF1A, HIN1 and APC) were highly methylated in tumors (82.5%, 75% and 52.5%, respectively) and adjacent normal tissues (85.2%, 70.4% and 44.4%, respectively). In order to determine concordance between gene methylation in tumors and adjacent normal-appearing tissues, the pair-wise agreement was estimated for each marker. As shown in Table II, hypermethylation of HIN1, RASSF1A, APC and TWIST1 in tumor and adjacent tissues were highly concordant (p=0.04, p=0.02, p=0.005 and p<0.0001, respectively, Fisher's exact test).

Map of gene promoter methylation in blood, normal adjacent and tumor tissues. Box color represents the degree of methylation (light gray, 1 to<4%; dark gray, 4 to<10%; black, ≥10%.
Promoter hypermethylation in WBC DNA in cases and controls. Of the eight tumor suppressor genes tested, BRCA1, HIN1, RASSF1A, CDH1 and RARβ were rarely hypermethylated in blood DNAs from the cases and their methylation levels were below 4% while APC, TWIST1 and CYCLIND2 were never hypermethylated limiting data analysis. Although there were limited data, most patients with WBC DNA methylation of BRCA1, HIN1, RASSF1A and CDH1 also had hypermethylation of those genes in tumor tissues. Moreover, for HIN1 and RASSF1A, the patients with WBC DNA methylation in these genes were also hypermethylated in adjacent tissues (Table II). There was no significant difference in WBC gene-specific methylation between cases and controls (Figure 1). We then investigated whether using possible combinations of genes which each had a higher frequency of methylation in cases compared to controls would show stronger associations with breast cancer. Hypermethylation in the panel of genes tested was associated with up to 2-fold increased breast cancer risk but the associations were not statistically significant (data not shown).
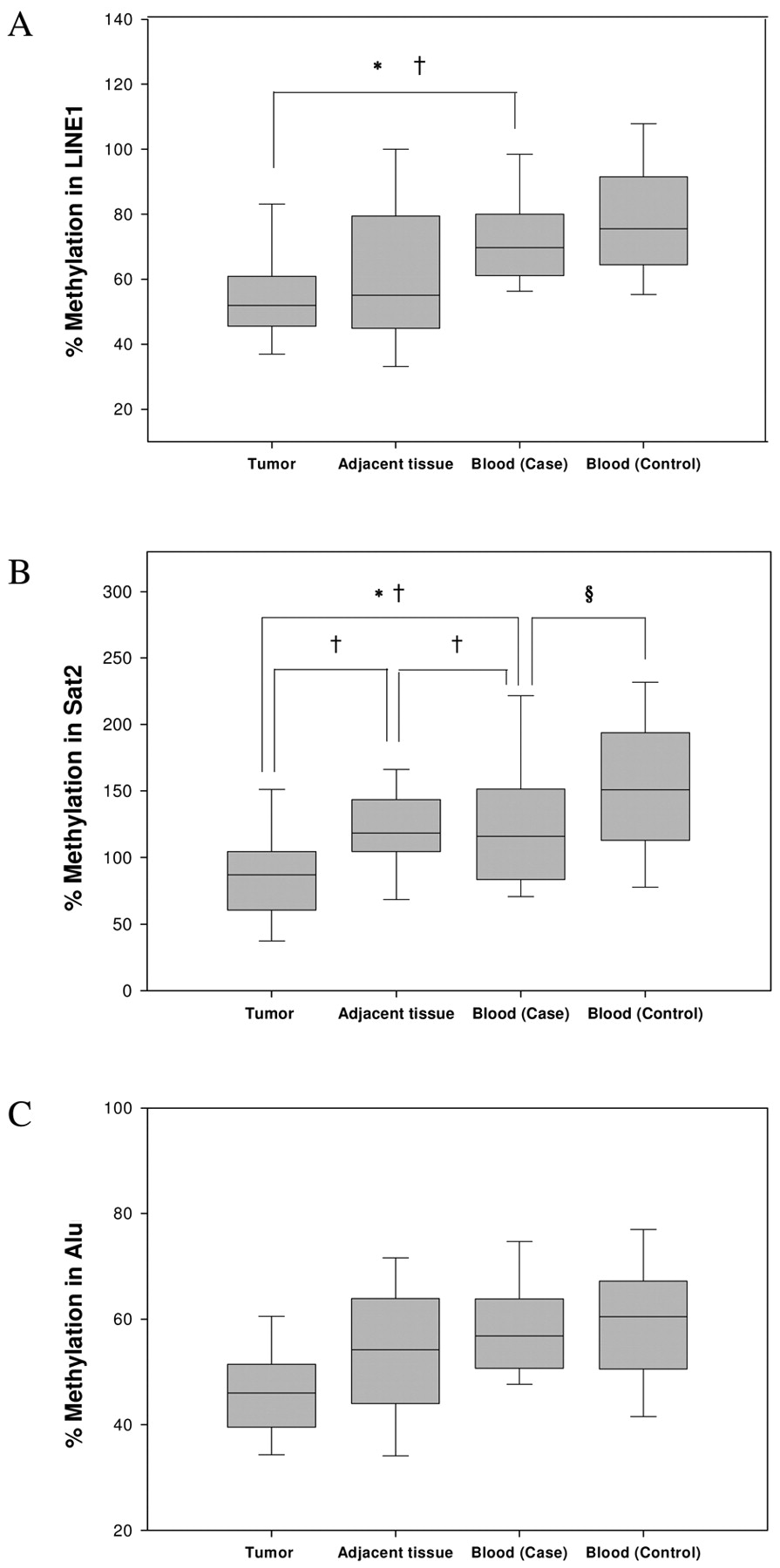
Hypomethylation of repetitive elements. Three repetitive elements, LINE1, Sat2 and Alu, were used as surrogates for measurement of genomic DNA hypomethy-lation. As shown in Figure 2, DNA methylation for the three repetitive elements was lower in tumor tissue compared to adjacent tissue and WBC DNA, but only that for Sat2 was significant. Significant correlations between methylation of Sat2 in tumor and adjacent normal tissues or adjacent normal tissue and WBC DNA were found (Rho=0.78; p<0.0001, Rho=0.67; p=0.002, respectively, Spearman's test, data not shown). Interestingly, methylation of LINE1 and Sat2 in tumor tissue was significantly lower compared to WBC DNA (both p<0.0001, Wilcoxon test) and correlated with WBC DNA (Rho=0.46; p=0.003, Rho=0.32; p=0.046, respectively, Spearman's test). Moreover, methylation of Sat2 in WBC DNA was significantly different between cases and controls after age adjustment (p=0.01, ANOVA test). The correlation between methylation of the three repetitive elements and clinicopathological parameters was further analyzed. Despite the limited data due to the small sample size and missing information, LINE1 in WBC showed significantly lower methylation levels in premeno-pausal women compared to postmenopausal (p=0.001, Wilcoxon test) (data not shown).

Comparison of (A) LINE1, (B) Sat2 and (C) Alu hypomethylation levels from tumor (n=40), normal adjacent tissues (n=27) and WBC DNA (n=40 for both cases and controls). Hypomethylation levels in LINE1 and Sat2 in tumor tissue was significantly decreased compared with those in WBC DNA (*both P<0.0001, Wilcoxon test). Significant correlations in methylation of LINE1 between tumor and WBC DNA (†Rho=0.46, P=0.0031, Spearman's rank correlation test) and methylation of Sat2 between tumor and adjacent normal tissues (†Rho=0.78, P<0.0001), tumor and WBC DNA (†Rho=0.32, P=0.046) or adjacent normal tissue and WBC DNA (†Rho=0.67, P=0.002) were shown. Methylation of Sat2 in WBC DNA was significantly different between cases and control (§P=0.01, Wilcoxon test). Data represent the means±SD (error bars).
Discussion
The present study examined methylation changes in tumor, adjacent normal tissues and WBC DNA from invasive ductal breast cancer patients to identify epigenetic markers of breast cancer risk. Hypermethylation of eight known tumorsuppressor genes were frequent in tumor and adjacent normal breast tissues but with lower levels in normal tissues than their paired-tumor tissue. Moreover, hypermethylation of APC, RASSF1A, HIN1 and TWIST1 were positively correla-ted between tumor and adjacent normal breast tissue. The presence of the aberrant methylation in tumor adjacent normal tissue might be due to disseminated cancer cells which have escaped routine pathological detection with immunostaining (41, 42). Field effects, shown in lung cancer, could also explain the aberrant methylation in normal tissue. Guo et al. (43) reported aberrant hypermethylation in multiple genes in histologically negative bronchial margins of resected non-small cell lung carcinomas which represents a field defect of widespread epigenetic change that occurs early in lung cancer. This observation suggests that the pattern of methylation in adjacent normal breast tissue DNA may be an important predictor of breast cancer risk. However, lack of methylation levels of those genes in normal breast tissue from controls limits our conclusion.
Although there were limited data, most cases with hypermethylation of BRCA1, HIN1, RASSF1A and CDH1 in blood also had hypermethylation of these genes in tumor tissues. This observation suggests that methylation of specific tumor suppressor genes in the blood could be a possible marker for breast cancer risk. Gene-specific methylation in WBC, however, did not differ between cases and controls. Further analyses combining data on genes with more frequent methylation in cases than controls were carried out to determine the association with breast cancer risk. While an increased risk was found for subjects with elevated WBC methylation in the possible gene panels, the difference was not statistically significant. There is preliminary evidence from several small studies that peripheral blood DNA contains epigenetic information, which may be a valuable predictive marker of an individual's risk of developing cancer (44-46). In contrast to our results, Snell et al. (47) reported that a low level of BRCA1 promoter methylation occurs in normal tissues of the body and is associated with the development of BRCA1-like breast cancer. The exact mechanism for the presence of aberrant methylation in both tumorous and non-tumorous DNA is not fully understood. Although studies in patients with breast (47) and prostate cancer (48) found higher tumor suppressor gene methylation in blood compared with controls, it is still not clear to what extent disseminated tumor cells contributed to this effect. However, it is unlikely that the MethyLight assay we used is sufficiently sensitive to detect the rare tumor cells that might be present in the WBC pellet. Meanwhile, the tendency for an association of methylation in the gene panel with increased breast cancer risk could be spurious or biased due to the small sample size, a major limitation of our study. Thus, further studies with larger sample sizes and investigation of additional genes are required in order to determine the relationship between DNA methylation in tumor and blood.
We also examined genomic hypomethylation by PCR analysis of three repetitive DNA elements, LINE1, Sat2 and Alu. Methylation of Sat2 in WBC DNA was significantly correlated with that in paired tumor and adjacent normal tissues from breast cancer patients. Moreover, methylation of Sat2 in WBC DNA was significantly different between cases and controls, while methylation levels of LINE1 and Alu in blood did not differ between the two groups. Sat2, a satellite DNA sequence is found predominantly in juxtacentromeric hetero-chromatin of certain human chromosomes (30). Sat2 hypomethylation may not be reflected in the total methylation content, since it makes up a minor fraction of repetitive DNA sequences. Recently, however, Lee et al. (49) reported that Sat2 hypomethylation occurred earlier than LINE1 or ALU hypomethylation along the multistep process of hepatocarcinogenesis. This finding suggested that Sat2 hypomethylation might be a surrogate biomarker for determination of early stage hepatocellular carcinoma. To date, very few studies reported an association between Sat2 methylation levels and risk of breast or ovarian cancers (26, 50). Our data show a positive association between methylation of Sat2 in WBC DNA and breast cancer risk, but further larger studies are needed to support this conclusion. In addition, several studies have reported decreased genomic methylation in blood DNA as a marker for neoplasia (34, 51, 52). A lower methylation level of LINE1 in WBC DNA was found in head and neck squamous carcinoma cases compared to controls (36). However, a study by Choi et al. (53) is in agreement with our results on LINE1 showing no difference in blood DNA methylation between breast cancer cases and controls.
Limitations of this study are the relatively small sample size and the lack of information on general demographic and clinicopathologic variables for some subjects. In addition, tissue was not available from controls. The small sample size and relatively modest number of genes tested precluded further statistical analysis of the possible relationship between the methylation levels and other life-style or clinicopathologic factors. Therefore, a larger sample size and investigation of more tumor-suppressor genes in future studies should clarify the role of methylation in breast carcinogenesis. Despite of these limitations, however, the strength of this study is the investigation of methylation of specific genes and repetitive elements in tumor, adjacent normal tissue and WBC DNA from the same individuals.
In summary, the present study showed that methylation of Sat2 in WBC DNA may be a surrogate biomarker for the determination of breast cancer risk. These findings could lead to simple and accessible measures of DNA methylation to help identify women at high risk for breast cancer. But larger studies of longitudinal nature are needed to establish the usefulness of WBC DNA methylation as a marker of risk.
Acknowledgements
This work was supported by an award from the Breast Cancer Research Foundation, NIH grants ES009089 and CA013696. Dr. Yazici was the recipient of an Avon Foundation-AACR International Scholar Award in Breast Cancer Research.
Footnotes
-
↵* These two authors are contributed equally to this work.
- Received March 17, 2010.
- Revision received May 31, 2010.
- Accepted June 4, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Aberrant epigenetic and transcriptional events associated with breast cancer risk
- LINE-1 Hypomethylation Is Associated With Malignant Traits and Cell Proliferation in Lung Adenocarcinoma
- Diagnostic value of RASSF1A methylation for breast cancer: a meta-analysis
- Whole-Blood DNA Methylation Markers in Early Detection of Breast Cancer: A Systematic Literature Review
- Physical Activity, Global DNA Methylation, and Breast Cancer Risk: A Systematic Literature Review and Meta-analysis
- Genomic DNA Hypomethylation and Risk of Renal Cell Carcinoma: A Case-Control Study
- DNA Methylation in Paired Breast Epithelial and White Blood Cells from Women Undergoing Reduction Mammoplasty
- Identification of Breast Cancer DNA Methylation Markers Optimized for Fine-Needle Aspiration Samples
- Is There a Link Between Genome-Wide Hypomethylation in Blood and Cancer Risk?
- Intragenic ATM Methylation in Peripheral Blood DNA as a Biomarker of Breast Cancer Risk
- Prenatal Smoke Exposure and Genomic DNA Methylation in a Multiethnic Birth Cohort