


Abstract
Curcumin (diferuloylmethane), which has no discernible toxicity, inhibits initiation, promotion and progression of carcinogenesis. 5-Fluorouracil (5-FU) or 5-FU plus oxaliplatin (FOLFOX) remains the backbone of colorectal cancer chemotherapeutics, but produces an incomplete response resulting in survival of cells (chemo-surviving cells) that may lead to cancer recurrence. The present investigation was, therefore, undertaken to examine whether addition of curcumin to FOLFOX is a superior therapeutic strategy for chemo-surviving cells. Forty-eight-hour treatment of colon cancer HCT-116 and HT-29 cells with FOLFOX resulted in 60-70% survival, accompanied by a marked activation of insulin like growth factor-1 receptor (IGF-1R) and minor to moderate increase in epidermal growth factor receptor (EGFR), v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (HER-2) as well as v-akt murine thymoma viral oncogene homolog 1 (AKT), cyclooxygenase-2 (COX-2) and cyclin-D1. However, inclusion of curcumin to continued FOLFOX treatment for another 48 h greatly reduced the survival of these cells, accompanied by a concomitant reduction in activation of EGFR, HER-2, IGF-1R and AKT, as well as expression of COX-2 and cyclin-D1. More importantly, EGFR tyrosine kinase inhibitor gefitinib or attenuation of IGF-1R expression by the corresponding si-RNA caused a 30-60% growth inhibition of chemo-surviving HCT-116 cells. However, curcumin alone was found to be more effective than both gefitinib and IGF-1R si-RNA mediated growth inhibition of chemo-surviving HCT-116 cells and addition of FOLFOX to curcumin did not increase the growth inhibitory effect of curcumin. Our data suggest that inclusion of curcumin in conventional chemotherapeutic regimens could be an effective strategy to prevent the emergence of chemoresistant colon cancer cells.
Despite recent incorporation of novel cytotoxic chemotherapy and biological agents in the treatment of metastatic colorectal cancer, the outcome remains poor (1, 2). Currently, 5-fluorouracil (5-FU) or 5-FU plus oxaliplatin (FOLFOX) remains the backbone of colorectal cancer chemotherapeutics. Although chemotherapeutic regimens containing FOLFOX produce a response in the majority of the cases, virtually all the responses are incomplete and emergence of resistance, with subsequent recurrence of the cancer, is universal.
Accumulating evidence suggests that the development and progression of many malignancies, including colorectal cancer, are associated with constitutive activation of multiple signaling pathways that promote proliferation, inhibit apoptosis and induce metastasis (3). A large body of evidence suggests that EGFR and/or its family members, specifically ErbB-2/HER-2 and ErbB-3/HER-3 (collectively referred to as EGFRs) play a crucial role in regulating several pathways that affect tumor cell survival, angiogenesis, motility and invasiveness (4-6). Insulin-like growth factor (IGF)/IGF-1R system has also been implicated in the development and progression of colorectal cancer (7, 8). In addition, EGFRs have been shown to mediate resistance to chemotherapeutic agents (9-11). Therefore, agent(s) that would target EGFRs and IGF-1R are likely to affect multiple aspects of tumor survival and chemoresistance.
Curcumin (diferuloylmethane), the major active ingredient of turmeric (Curcuma longa) with no discernable toxicity, has been shown to inhibit the growth of transformed cells and colon carcinogenesis at the initiation, promotion and progression stages in carcinogen-induced rodent models (12-16). Curcumin has also been shown to prevent the development of adenomas in the intestinal tract of Min+/− mice, a model of human familial adenomatous polyposis (17). In a phase I clinical trial, curcumin has been found to be effective in inhibiting the growth of a variety of tumors (18). Recently, we reported that curcumin synergizes with FOLFOX by inhibiting all EGFRs as well as IGF-1R in colon cancer cells (19).
However, it remains to be determined whether curcumin would be effective against chemo-surviving cells, the cells that survive acute chemotherapy insult. The current in vitro study was undertaken to examine the effect of curcumin on the growth of chemo-surviving colon cancer cells (cells that survived 48-h treatment with FOLFOX). We have also examined the possible mechanism of growth inhibition focusing on the regulation of growth factor receptors pathways, specifically EGFRs and IGF-1R which are considered to play an important role in the progression of colorectal cancer.
Materials and Methods
Cell lines and cell cultures. Human colon cancer HCT-116 and HT-29 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) in tissue culture flasks in a humidified incubator at 37°C in an atmosphere of 95% air and 5% CO2. The medium was supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic/anti-mycotic andwas changed three times a week. Cells were passaged using trypsin/EDTA. Curcumin was purchased from Sigma Chemical Co. (St. Louis, MO, USA) and dissolved in 100% ethanol and subsequently diluted with the tissue culture medium.
Growth inhibition assay. Inhibition of cell growth in response to curcumin and/or FOLFOX was assessed by 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously (15, 24). Briefly, cells were dispersed by trypsin-EDTA treatment and 2.5×104 cells/ml, re-suspended in DMEM containing 10% FBS and seeded into 96-wells culture plates with 6 replicates. After 24 h of plating, incubation was continued for another 48 h in the absence (control) or presence of FOLFOX alone and in combination with 25 μM or 50 μM of curcumin. At the end of the 48 h incubation period, the reaction was terminated by adding 20 μl of 5 mg/ml stock of MTT to each well. The reaction was allowed to proceed for 3-4 hours at 37°C. The formazan crystals were then dissolved by adding 0.1 ml of dimethyl sulfoxide (DMSO). The intensity of the color developed, which is the reflection of number of live cells, was measured at a wavelength of 570 nm. All values were compared to the corresponding controls. All assays were performed with 6 replicates.
Chemo-surviving cell preparation. HCT-116 and HT-29 cells grown in monolayer were incubated with 50 μM 5-FU and 1.25 μM oxaliplatin (FOLFOX) (both 5-FU and oxaliplatin were obtained from Sigma-Aldrich, St. Louis, MO, USA) for 48 h, subsequently washed twice with phosphate-buffered saline (PBS). The cells that remained attached to the plate following the PBS wash (referred to as chemo-surviving cells) were trypsinized and regrown as a monolayer. Subsequent experiments of growth inhibition with gefitinib, curcumin and or FOLFOX were performed on these regrown chemo-surviving cells.
Western blot analysis. Western blot analysis was performed essentially according to our standard protocol (19). Briefly, the cells were solubilized in lysis buffer [50 mM Tris; 100 mM NaCl; 2.5 mM EDTA; 1% Triton X-100; 1% Nonidet P-40; 2.5 mM Na3VO4; 25 μg/ml aprotinin; 25 μg/ml leupeptin; 25 μg/ml pepstatin A; and 1 mM phenylmethylsulfonyl fluoride (PMSF)]). Following clarification at 10,000 ×g for 15 min, the supernatant was used for Westernblot analysis. In all analyses, protein concentration, determined by the Bio-Rad Protein Assay kit (Bio-Rad, Hercules, CA, USA), was standardized among the samples. Aliquots of cell lysates containing 50 μg of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Following electrophoresis, proteins were transferred electrophoretically onto supported nitrocellulose membranes (Osmonics, Gloucester, MA, USA). Membranes were incubated for 1 h at room temperature with blocking buffer, TBS-T (20 mM Tris, pH 7.6, 100 nM NaCl, 0.1% Tween-20) and 5% nonfat dry milk with gentle agitation. After washing the membranes with TBS-T, they were incubated overnight at 4°C in TBS-T buffer containing 5% milk and with one of the following antibodies (1:1000 dilution): phospho-EGFR (Tyr1173), phospho-ErbB-2/HER-2 (Tyr1121), phospho-IGF-1R, phospho-Akt (Ser473) or COX-2 (Cell Signaling, Boston, MA, USA). The membranes were washed 3 times with TBS-T, and subsequently incubated with appropriate secondary antibodies (1:5000 dilutions) (Santacruz Biotechnology, Santa Cruz, CA, USA) in TBS-T containing 5% milk for 2 hours at room temperature with gentle agitation. The membranes were washed again with TBS-T, and the protein bands were visualized by enhanced chemiluminescence (ECL) detection system (Amersham, Sweden). The membranes containing the electrophoresed proteins were exposed to X-Omat film (PerkinElmer, Waltham, MA, USA). The membranes were then stripped (2× for 15 min at 55°C) in stripping buffer containing 100 mM 2-mercaptoethanol, 2% SDS and 62.5 mM Tris-HCl pH6.7, and reprobed with β-actin as loading control. All Western blots were performed at least three times for each experiment. Densitometric measurements of the scanned bands were performed using the digitized scientific software program UN-SCNAT. Data were normalized to β-actin.
Transfection with si-RNAs. Chemo-surviving HCT-116 cells were transfected with the vector plasmid, EGFR si-RNA or IGF-1R si-RNA (Ambion, Austin, TX, USA) using Lipofectamine 2000 according to the manufacturer's instruction. For transfection, approximately 0.5-1×106 HT-29 or HCT-116 cells in 2 ml DMEM/10% FBS were plated in 96-well tissue culture dishes. After 24 h, the cells were transfected with Lipofectamine-2000 according to the manufacturer's instruction. Two micrograms of vector, EGFR si-RNA or IGF-1R si-RNA were used in each well, and incubated for 48 h.
Statistical analysis. Data are represented as the mean±SEM. The statistical significance of differential findings between experimental groups and control was determined by Student's t-test as implemented by Excel 2000 (Microsoft Corp., Redmond, WA, USA). P-values smaller than 0.05 were considered statistically significant.
Results
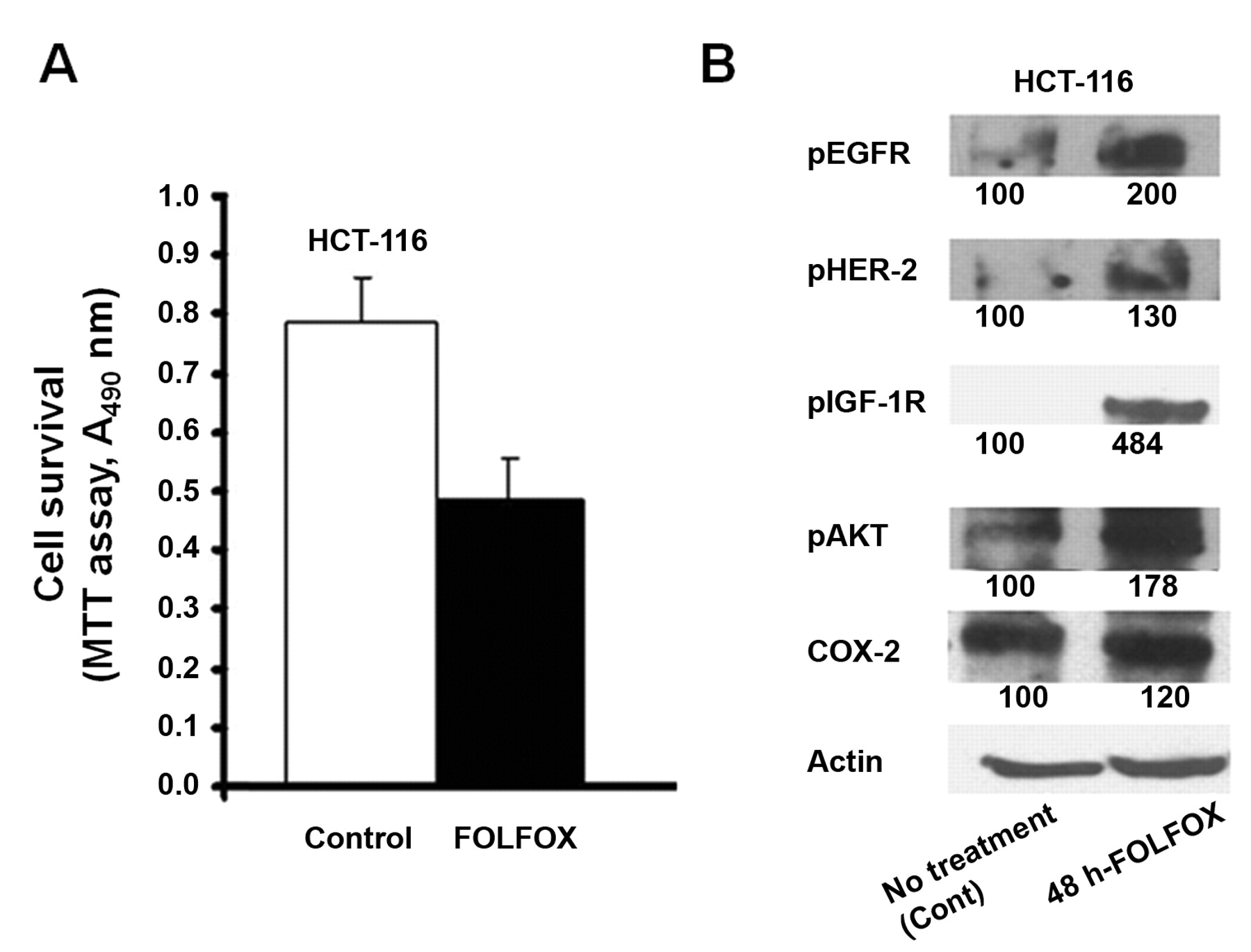
An MTT assay was performed to determine the survival rate of colon cancer HCT-116 and HT-29 cells following FOLFOX treatment. We observed that 60-70% of the cells survived following 48 h incubation with FOLFOX (50 μM 5-FU and 1.25 μM oxaliplatin), henceforth referred to as chemo-surviving cells (Figure 1A data for HT-29 are not shown as they were similar to HCT-116).

A: Proportion of chemo-surviving HCT-116 cells following 48 h of treatment with FOLFOX (50 μM 5-FU and 1.25 μM oxaliplatin) chemotherapy, as measured by MTT assay. B: Western-blot showing the levels of activated (tyrosine phosphorylated) forms of EGFR, HER-2, IGF-1R, and downstream mediators AKT, as well as COX-2, in chemo-surviving HCT-116 cells.
Since members of EGFR family and IGF-1R play an important role in development and progression of colorectal cancer (5-8), we postulated that the chemo-surviving cells would exhibit increased expression and/or activation of survival promoting growth factor receptors, specifically EGFR and its family members and IGF-1R. Indeed, we observed a robust a 2-fold and 4.8-fold increase in activation of EGFR and IGF-1R, respectively, in chemo-surviving HCT-116 cells (Figure 1B) compared to untreated cells (control). There was also a modest 30% increase in HER2 activation (Figure 1B). We also examined the expression and activation of AKT and COX-2, both of which are known to be critically involved in regulating cell survival signaling, downstream of growth factor receptors in various types of cancer including colorectal cancer. We observed a 20% increase in expression of COX-2 and a 78% increased activation of AKT without a significant change in its expression, when compared with the untreated parental cell controls (Figure 1B).
On the basis of our previous observation that curcumin synergizes with FOLFOX through down-regulation of EGFRs and IGF-1R, we hypothesized that curcumin would cause growth inhibition of chemo-surviving colon cancer cells. To test this hypothesis, chemo-surviving HCT-116 and HT-29 cells were subjected to further treatment for 48 h with FOLFOX or FOLFOX with two different doses of curcumin (25 or 50 μM). Incubation with the growth media only served as controls. Parental HCT-116 and HT-29 cells were also included to show changes in cell survival after 48 h FOLFOX treatment. Treatment of FOLFOX for 48 h resulted in approximately 50% and 60% survival of HCT-116 and HT-29 cells, respectively, when compared with the corresponding untreated parental cells. Further treatment of chemo-surviving HCT-116 and HT-29 cells with FOLFOX produced only a very modest growth inhibition (16-27%), when compared with the media-treated controls (Figure 2A). However, treatment of the chemo-surviving HCT-116 and HT-29 cells with FOLFOX together with either dose of curcumin exhibited significant growth inhibition compared to the media-treated controls (Figure 2A): low-dose curcumin (25 μM) in combination with FOLFOX was highly effective, producing a 60% growth inhibition in both HCT-116 and HT-29 chemo-surviving cells, and inhibition increased to 70-78% with the 50 μM dose of curcumin (Figure 2A).
Experiments were then performed to examine the effect of FOLFOX alone and in combination with curcumin (25 μM) on activation of the growth factor receptors and downstream mediators that are increased in chemo-surviving cells. Since curcumin was found to be effective in inhibiting growth of chemo-surviving colon cancer HCT-116 cells, whereas FOLFOX was not, we hypothesized that curcumin alone or in combination with FOLFOX would result in attenuation of growth factor signaling that is upregulated in chemo-surviving cells. Indeed, continued treatment of chemo-surviving HCT-116 cells with FOLFOX resulted in either no change or only a slight increase in activation of EGFR, HER2 and IGF-1R as well as downstream signaling molecules AKT and COX-2 compared to the media-treated controls (Figure 2B). On the other hand, addition of curcumin to FOLFOX resulted in a marked 39-74% inhibition of activation of EGFR, HER2 and IGF-1R (Figure 2B). Additionally, there was a 91% reduction in the expression of COX-2 and a 36% attenuation in activation of AKT with the combined treatment of curcumin and FOLFOX, compared to the media-treated controls (Figure 2B).

A: Growth inhibition (MTT assay) of chemo-surviving HCT-116 and HT-29 cells (pre treated with FOLFOX for 48 hours) with FOLFOX alone or in combination with curcumin (25 and 50 μM), compared to media-treated cells (control) following additional 48 h treatment. Untreated parental cells are included for purposes of comparison (total of 96 hours' exposure to media alone). B: Western blot showing the levels of activated growth factor receptors EGFR, HER-2, IGF-1R, and downstream mediators AKT, as well as COX-2, in chemo-surviving HCT-116 cells following treatment with FOLFOX alone or in combination with curcumin (25 and 50 μM), compared to media-treated cells (control). Parental untreated cells were included for purposes of comparison (total of 96 hours exposure to media alone). *p<0.001 Compared to control.
Since EGFR activation is increased in chemo-surviving cells and is attenuated by curcumin, we sought to determine whether and to what extent the selective EGFR tyrosine kinase inhibitor gefitinib (Iressa) would be effective in growth inhibition of chemo-surviving HCT-116 colon cancer cells. Pretreatment of chemo-surviving HCT-116 cells with gefitinib (1 μM) for 24 hours caused a 40% growth inhibition (Figure 3). The gefitinib-pretreated cells were then washed and subjected to treatment with the media alone (control), gefitinib, curcumin or curcumin plus FOLFOX for 48 h. Treatment with the media alone resulted in regrowth of chemo-surviving cells, whereas continued gefitinib treatment caused a 60% growth inhibition at the end of the 3 day experimental period (including a 24-h pretreatment period) (Figure 3). However, treatment with curcumin alone or curcumin and FOLFOX resulted in a marked >90% growth inhibition (Figure 3). It is noteworthy that addition of chemotherapy to curcumin resulted in no additional benefit over that observed with curcumin alone in inhibiting the growth of chemo-surviving colon cancer HCT-116 cells (Figure 3).
We then examined the effect of individual inhibition of EGFR or IGF-1R on growth inhibition of chemo-surviving HCT-116 cells. Transfection of chemosurviving HCT-116 cells with EGFR or IGF-1R si-RNA resulted in statistically significant 30% growth inhibition (Figure 4). Curcumin, which we have shown to inhibit EGFR as well as IGF-1R (19), caused a robust 70% growth inhibition at 48 h (Figure 4). Addition of curcumin to EGFR-si RNA and IGF-1R si-RNA transfected cells resulted in a minor further growth inhibition compared to curcumin alone (Figure 4).
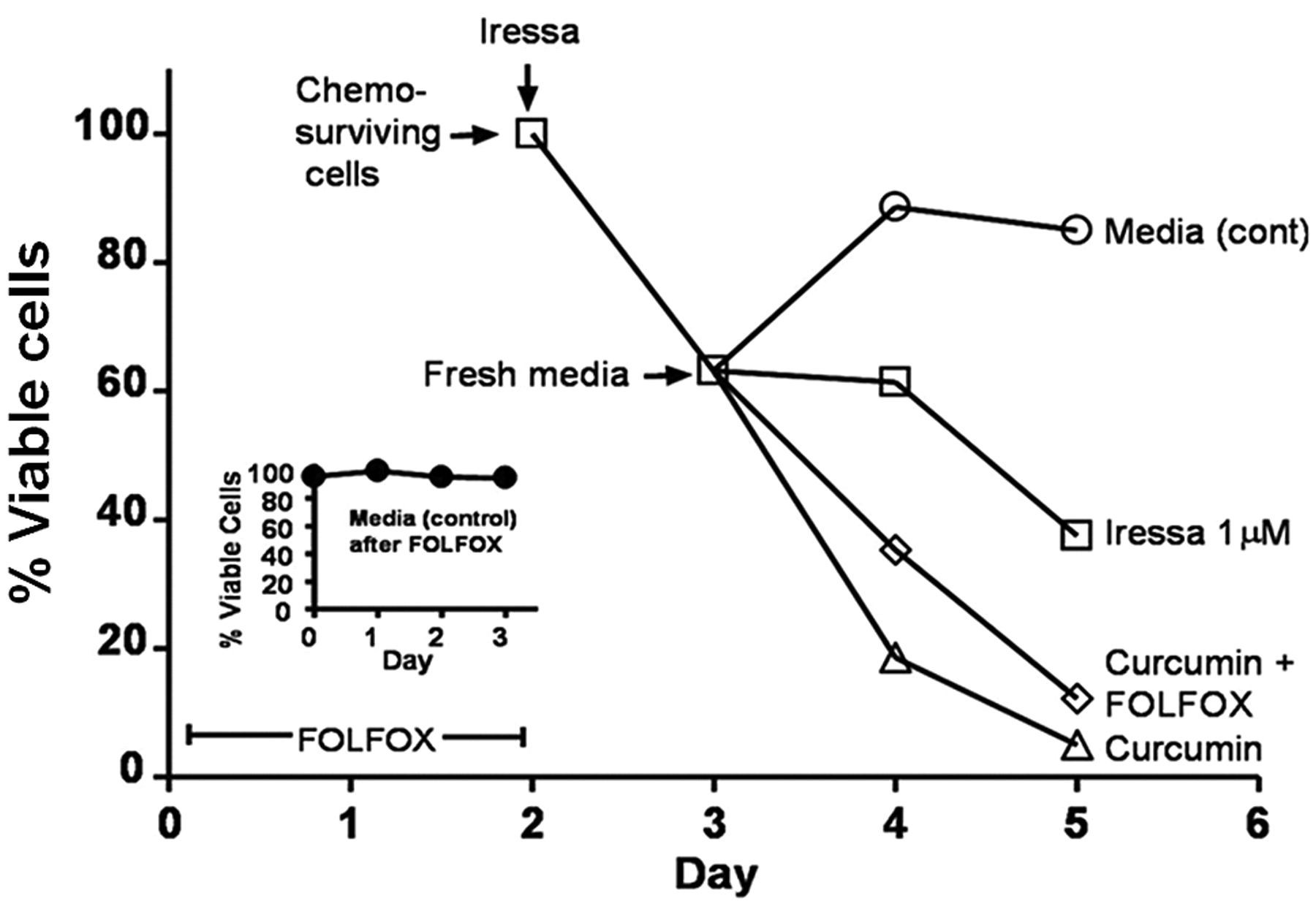
Growth inhibition (MTT assay) of chemo-surviving HCT-116 cells pretreated with 1 μM gefitinib followed by additional 48 h treatment with gefitinib, curcumin or FOLFOX and curcumin compared to cells in media alone (control).
Discussion
Chemotherapy produces incomplete responses in a vast majority of cancer cases particularly of colorectal cancer (2). This leads to survival of a population of cells within the tumor resulting in subsequent chemotherapy-resistant relapses. The precise mechanism of this phenomenon of chemo-survival remains unknown.
Abnormal activity of EGFRs has been associated with the development and progression of many malignancies, including that of the colon. In particular, overexpression of EGFR and HER2 in colorectal cancer correlates with an extremely poor clinical prognosis (20, 21). The majority of solid tumors, including those of the colon, overexpress one or more members of the EGFR family and coexpression of EGFR with HER-2 or HER-3 results in the development of enhanced drug resistance (9, 22). More recently, IGF-1R is also emerging as an important pathway responsible for the development and progression of colorectal cancer. In addition, there is crosstalk between EGFRs and IGF-1R, resulting in therapeutic resistance with targeting individual pathways (23). EGFR inhibitors have been successfully incorporated in the therapeutic armamentarium of colon cancer. However, the benefits appear to be modest and the complete responses are rare (24). One of the mechanisms of resistance to EGFR inhibitors is their hetrodimerization with IGF-1R receptor (25). Hence, co-targeting of EGFR and IGF-1R would likely result in greater therapeutic efficacy. In our model of chemo-surviving colon cancer cells, we show that not only EGFR and its family members, but also IGF-1R, are significantly activated, confirming their involvement in survival of these cells. Therefore, targeting them may provide beneficial effect in terms of reversing chemotherapy resistance. Recently a report by Dallas et al., who demonstrated inhibition of growth of oxaliplatin-resistant colon cancer cells by inhibiting IGF-1R, further supports the contention that certain growth factor receptors play a critical role in cells surviving chemotherapy insult (11). However, one of the major concerns with combining various biological agents is an increase in overall toxicity. Hence, the development of pleiotropic agents with minimal toxicity is highly desirable in combating the emergence of chemo-surviving cells and the resultant subsequent relapse. Our current data demonstrate that one can target these survival pathways with non-toxic pleiotropic agents such as curcumin for therapeutic gains.

Growth inhibition (MTT assay) of chemo-surviving HCT-116 cells transfected with vector, EGFR si-RNA, IGF-1R si-RNA and treated with either DMSO (control) or curcumin. *p<0.01 and **p<0.001 compared to control.
Curcumin [diferuloylmethane; 1,7-bis-(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione], the major active ingredient in turmeric powder, affects promotion/progression phases of colon carcinogenesis (12-16). The effect of curcumin on progression of the cancer is pleiotropic. A plethora of evidence suggests that the chemopreventive effect is mainly due to inhibition of COX-2, which leads to inhibition of prostaglandin E and of subsequent colon cancer growth (9, 26). COX-2 inhibition is due to attenuation of activation of transcription factors activator protein-1 (AP-1) and nuclear factor-κB (NF-κB), which regulate its transcription (26, 27). There is some evidence that curcumin inhibits the ligand-induced activation of EGFR and reduces its transcription via inhibition of erg-1 transcription factor (28). Moreover, our recent data demonstrate that curcumin inhibits both expression and activation of not only EGFR but also its family members and IGF-1R (19). In this report, we show that curcumin is not only highly effective in inhibiting the growth of chemo-surviving colon cancer cells but is also superior to EGFR inhibition by gefitinib and to IGF-1R attenuation through IGF-1R si-RNA. We believe that the ability of curcumin to effectively downregulate multiple growth factor receptor-mediated survival signals is critical in inhibiting chemo-surviving colon cancer cells. Curcumin has been shown to have no discernible toxicity in multiple human studies. Hence, it would be an ideal agent to use in combination with chemotherapy to prevent the emergence of chemo-surviving cells.
In conclusion, chemo-surviving colon cancer cells pose a major threat to the success of colon cancer chemotherapeutics via up-regulation of EGFRs and IGF-1R. Inclusion of curcumin, which effectively inhibits EGFRs and IGF-1R in chemo-surviving cells, into colon cancer chemotherapeutics is likely to inhibit the growth of the chemo-surviving cells and reduce the likelihood of subsequent relapse following chemotherapy at no additional toxicity.
Acknowledgements
The work was supported by grants from the National Institutes of Health/National Institute on Aging (APNM; R01 AG014343), Department of Veterans Affairs.
Footnotes
- Received August 21, 2009.
- Revision received January 4, 2010.
- Accepted January 5, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A Unique Nonsaccharide Mimetic of Heparin Hexasaccharide Inhibits Colon Cancer Stem Cells via p38 MAP Kinase Activation
- Curcumin mediates anticancer effects by modulating multiple cell signaling pathways
- The Molecular Basis for the Pharmacokinetics and Pharmacodynamics of Curcumin and Its Metabolites in Relation to Cancer
- Novel Curcumin Oral Delivery Systems
- Death by Design: Where Curcumin Sensitizes Drug-resistant Tumours