


Abstract
Background: Dendritic cells (DCs) are the professional antigen-presenting cells of the immune system. We have demonstrated that vaccination of autologous ex vivo cultured DCs results in the induction of tumor-specific immune responses in cancer patients, which correlates with clinical response. Optimization of antigen loading is one of the possibilities for further improving the efficacy of DC vaccination. Theoretically, transfection of DCs with RNA encoding a tumor-specific antigen may induce a broader immune response as compared to the most widely used technique of peptide pulsing. Patients and Methods: In this clinical study, RNA transfection was compared with peptide pulsing as an antigen loading strategy for DC vaccination. Patients with resectable liver metastases of colorectal cancer were vaccinated intravenously and intradermally 3 times weekly with either carcinoembryogenic antigen (CEA)-derived HLA-A2 binding peptide-loaded or CEA mRNA electroporated DCs prior to surgical resection of the metastases. All DCs were loaded with keyhole limpet hemocyanin (KLH) as a control protein. Evaluation of vaccine-induced immune reactivity consisted of T-cell proliferative responses and B-cell antibody responses against KLH in peripheral blood. CEA reactivity was determined in T-cell cultures of biopsies of post-treatment delayed type hypersensitivity skin tests. Results: Sixteen patients were included. All patients showed T-cell responses against KLH upon vaccination. CEA peptide-specific T-cells were detected in 8 out of 11 patients in the peptide group, but in none of the 5 patients in the RNA group. Conclusion: In our study, DC CEA mRNA transfection was not superior to DC CEA peptide pulsing in the induction of a tumor-specific immune response in colorectal cancer patients.
- Colorectal cancer
- dendritic cells
- antigen loading
- CEA peptide
- mRNA
- electroporation
- immunotherapy
- vaccination
Dendritic cells (DCs) are the most potent antigen-presenting cells, and are capable of inducing an effective antitumor immune response (1). Currently they are employed in clinical immunization protocols in cancer patients and objective clinical and immunological responses have been observed (2). However, a lot of crucial questions regarding the optimal preparation and administration of DC vaccines for clinical use remain unanswered to date (3). One of these questions concerns the optimal method of antigen loading. In the vast majority of studies, tumor antigenic peptides have been used (2). This has the advantage that the monitoring of the immune response is focused on a limited and predefined number of epitopes. However, pulsing DCs with peptides limits the response to identified epitopes and therefore limits the treated patient group to certain specific HLA types. In addition, peptide-pulsing does not account for post-translational modifications of the tumor antigens. These limitations may not exist for the expression of tumor antigens by DCs through RNA transfection, which has been shown effective in vitro and in vivo (4-6). Moreover, RNA transfection may lead to a more prolonged presentation of the antigen as compared to peptide-loading which appears to be short-lived (7). The feasibility, safety, and potential efficacy of non-virally RNA-transfected DCs has been shown in clinical studies with cancer patients (8-12). On the other hand, RNA transfection has potential drawbacks including a variable antigen expression and a low yield of viable cells after transfection, especially when using the technique of RNA electroporation (10). The true benefit of using RNA transfection over peptide loading should be derived from clinical trials. A clinical comparison of RNA-transfected DCs with peptide-pulsed DCs, however, has not been reported. Here, we compare the immunogenicity of an RNA-based DC vaccine versus a peptide-based DC vaccine in colorectal cancer patients. We included patients with resectable colorectal cancer liver metastases for the following reason. Vaccine therapy is likely to have greater efficacy in patients with low tumor volume compared to patients with widespread bulky metastases in whom immune suppression is often present. Patients with resected liver metastases have a high risk of relapse and may therefore potentially benefit from adjuvant treatment, such as vaccine therapy. Therefore, we opted to test the immunogenicity of our method in this specific subgroup of patients.
Patients and Methods
Study design. This was an open-label, single-institution, exploratory study in which monocyte-derived DCs loaded with CEA peptide or electroporated with CEA-encoding mRNA were administered to patients with resectable liver metastases of colorectal cancer. It was planned that the first 10 patients would receive DCs pulsed with CEA peptides, and another 10 patients would receive DCs transfected with mRNA. Approval from the local regulatory committee was obtained and the trial was registered at ClinicalTrials.gov (NCT00228189).
Objectives. The primary endpoint was to compare the immunogenicity of CEA peptide-loaded DCs with DCs electroporated with CEA mRNA. Secondary endpoints were the toxicity and the feasiblity of CEA-specific vaccination in colorectal cancer patients.
Patients. Inclusion criteria included: patients with liver metastases from CEA-expressing colorectal cancer scheduled for surgical resection, HLA-A0201 phenotype, Eastern Cooperative Oncology Group (ECOG) performance status 0-1, age ≥18 years, no clinical signs of extrahepatic metastases, no prior chemotherapy, immunotherapy or radiotherapy within 2 months before planned surgery.
DC preparation. DCs were generated either from buffy coats or from leukapheresis products. Monocytes were enriched from buffy coats as follows: Patients donated 0.5 l blood, from which a buffy-coat was made (Sanquin Bloodbank Nijmegen, the Netherlands). Peripheral blood mononuclear cells (PBMCs) were isolated by PureCell® (Medicult, Denmark) density gradient centrifugation (20 min, 20°C, 2100 rpm) and monocytes were enriched by adherence to plastic. Monocytes were enriched from leukapheresis products by counterflow elutriation using Elutra-cell separator (Gambro BCT, Inc, Hechingen, Germany) and single-use, functionally sealed disposable Elutra sets, as described elsewhere (14) and according to the manufacturer's instructions.
Monocytes were cultured in Cellgro® medium supplemented with interleukin (IL)-4 (500 units/ml) and granulocyte macrophage colony-stimulating factor (GM-CSF; 800 units/ml; all Cellgenix, Freiburg, Germany). Keyhole limpet hemocyanin (KLH; 10 μg/ml; Calbiochem, USA) was added at day 3 of culture, and 2 days before harvesting we added the maturation cocktail consisting of prostaglandin E2 (PGE2, 10 μg/ml; Pharmacia & Upjohn, Puurs, Belgium), tumor necrosis factor alpha (TNFα, 10 ng/ml), IL-1β (5 ng/ml) and IL-6 (15 ng/ml; all CellGenix). Release criteria were as previously described (3): more than 80% of the DC expressed high levels of CD80 and CD86. Mature DCs were electroporated with mRNA encoding CEA directly after harvesting as described below or pulsed with wild-type CEA peptide CAP-1 (CEA571-579, YLSGANLNL; Clinalfa, Switzerland) (13,15) directly after harvesting or after thawing (16, 17).
Electroporation of DC. Mature DCs were washed twice in phosphate-buffered solution (PBS) and once in OptiMEM without phenol red (Invitrogen, Breda, the Netherlands). Twenty micrograms of mRNA were transferred to a 4-mm cuvette (Bio-Rad, Veenendaal, the Netherlands) and 12×106 cells were added in 200 μl OptiMEM and incubated for 3 min before being pulsed in a Genepulser Xcell (Bio-Rad) by an exponential decay pulse of 300 V, 150 μF, as described elsewhere (10). Electroporation was carried out under good manufacturing practice conditions. For the first vaccination, 2 to 3 aliquots of 12×106 DCs were electroporated with CEA-encoding mRNA. Electroporation efficiency was analyzed by flow cytometry. DCs for the first vaccination were injected 4 h after electroporation. DCs for subsequent vaccinations were frozen 2 h after electroporation, thawed on the day of vaccination, and incubated for an additional 2 h at 37°C before injection.
Treatment schedule. Vaccinations were administered three times at day 0, 7 and 14. The patients received 5×106 DCs intradermally (i.d.) in the upper leg, 5-10 cm from an inguinal lymph node. The remaining cells were given intraveneously (i.v.) as a bolus injection at the same time point. On day 26, a post-treatment delayed type hypersensitivity (DTH) test was performed (see below). At day 28, 6 mm punch biopsies were taken from the post-treatment DTH reaction sites. PBMCs were obtained before and after vaccination.
The same schedule of 3 weekly i.v./i.d. vaccinations followed by a post-treatment DTH was repeated twice at intervals of 6 months in the absence of recurrent disease. Serum CEA levels were measured before and after completion of the vaccination protocol.
Immunologic monitoring. CD4+ T-cell responses against KLH were measured using a 3H-thymidine incorporation proliferation assay with PBMCs of the patients before and after vaccination (17). The index was calculated as the counts ratio between KLH-stimulated PBMCs and non-stimulated PBMCs. Antibodies against KLH were measured in the serum of vaccinated patients by enzyme-linked immunosorbent assays (ELISA) as previously described (17, 18).
Post-treatment DTH reactions were performed as described previously (19). Briefly, CEA peptide only (100 μg in 100 μl), DCs pulsed with CEA peptide, DCs pulsed with KLH and CEA peptide, and DCs pulsed with KLH only (0.4-5×105 DCs each in approximately 100-200 μl) were injected i.d., 5-10 cm from an inguinal lymph node at different sites, in the upper leg contralaterally from the leg in which the DC vaccinations were performed. The maximum diameter of induration was measured after 48 h. T-Cell culture from DTH biopsies was performed in low dose IL-2 (100 U/ml; Proleukin®, Chiron, the Netherlands) for approximately 2 weeks without ex vivo restimulation with antigen as described before (19). After 2 to 4 weeks of culturing, T-cells were tested for antigen recognition or tested for tetramer binding.
DTH-derived cells (1×105 cells in 10 μl) were incubated with Phycoerythrin (PE)-labeled CEA and cytomegalovirus (CMV) tetrameric-MHC complexes (Sanquin, Amsterdam, the Netherlands) for 60 min at room temperature. In the last 20 min of this incubation, fluorescein isothiocyanate (FITC)-conjugated monoclonal antibodies directed against CD8 (Becton Dickinson, Mountain View, CA, USA) were added. After washing, the samples were analyzed by flow cytometry. In all analyses CEA tetramer staining was compared with CMV tetramers as a negative control.
Vaccine-induced antigen-specific immune responses.
Production of cytokines by the total population of DTH-derived cells was measured in response to T2 cells pulsed with CEA peptide or an irrelevant HLA-A2.1 binding peptide (tyrosinase or G250) or in response to PBMCs electroporated with CEA mRNA. Cytokine production was measured in supernatants after 16 h by cytometric bead array (Th1/Th2 Cytokine CBA 1; BD Pharmingen, San Diego, CA, USA). IFN-γ production was considered positive when IFN-γ levels of DTH-infiltrating T-cells were more than 10-fold higher after co-incubation with CEA-loaded T2 cells compared with co-incubation with T2 cells loaded with irrelevant antigen.
Patient evaluation. Clinical follow-up consisted of history, physical examination, serum CEA level, CT scanning of the liver and X-ray of the chest at 3-month intervals.
Statistical analysis. Data were analyzed statistically by means of analysis of variance and Student-Newman-Keuls test, or by means of Mann-Whitney U nonparametric statistics. Statistical significance was defined as p<0.05.
Results
Patients. Sixteen patients were included in the study. In the period 2004-2005, eleven patients received peptide-pulsed DCs (previously reported in (13)). One extra patient was included as planned in case a patient proved not to be evaluable. Due to a slow accrual of only 5 patients in the second phase of the study, as well as the results in these patients, the study was prematurely closed. Baseline characteristics between patients receiving peptide-pulsed DCs and RNA-loaded DCs were comparable (data not shown) and none of the patients had been treated with chemotherapy.
Clinical outcome and toxicity. Four out of 11 patients in the peptide group and 1 out of 5 patients in the RNA group had irresectable disease at surgery. In the peptide-group, one patient (pep-5) was diagnosed with lung metastases before surgery and one patient died of postoperative complications not related to the vaccination treatment (pep-9). No toxicity other than grade 1 flu-like symptoms, fever and injection site reactions were observed in both groups. Specifically, no diarrhea or other signs of autoimmune colitis were observed. Seven patients treated with peptide-pulsed DCs and two patients treated with mRNA-loaded DCs showed a transient and modest rise in serum CEA level upon vaccination, defined as an increase of ≥15% compared with baseline (33). In the patients who underwent a radical resection of metastases, the median progression-free survival after treatment with peptide-pulsed DCs and mRNA-loaded DCs was 18 months (range 1-77 months) and 26 months (range 13-41 months), respectively.

DC characteristics. A: Expression of HLA-ABC, HLA-DR/DP, HLA-DQ, CD80, CD83, CD86 and CCR7 was measured by flow cytometry on mature DCs for the first i.v./i.d. vaccination. Results are shown as the mean±SEM of the percentage of cells expressing the maturation markers of peptide-loaded DCs (grey bars) and mRNA-electroporated DCs (black bars). B: CEA protein expression was measured by flow cytometry 2 h after electroporation with mRNA. Results are shown as the percentage of cells expressing CEA protein for each individual patient.
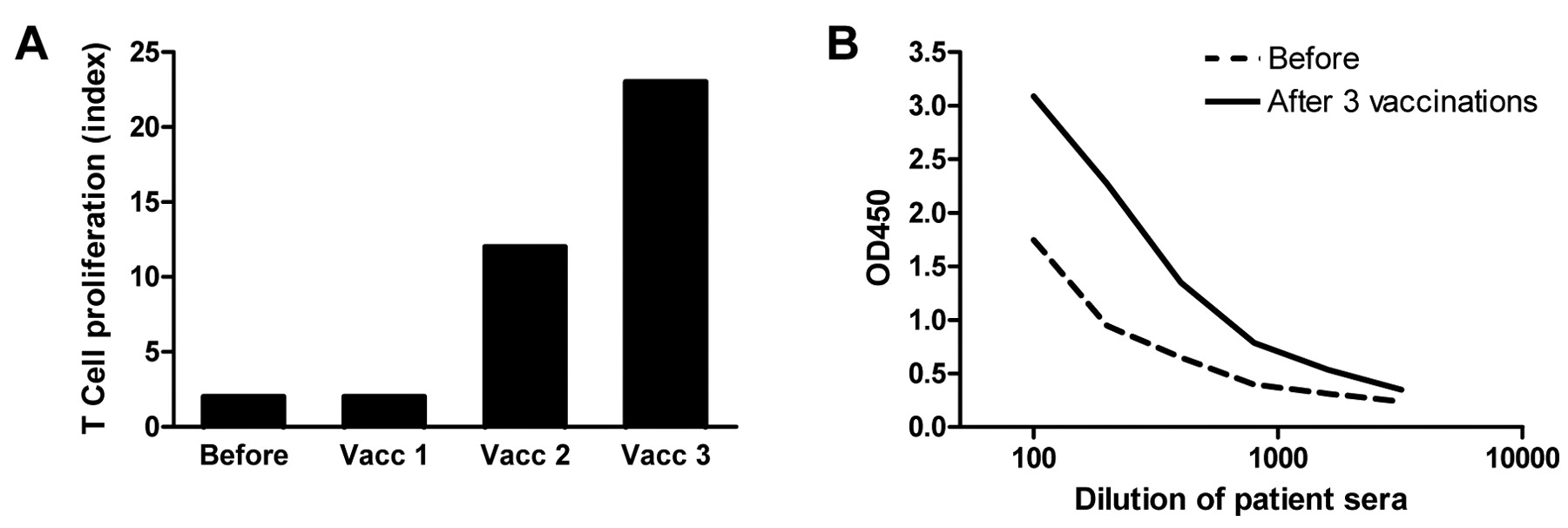
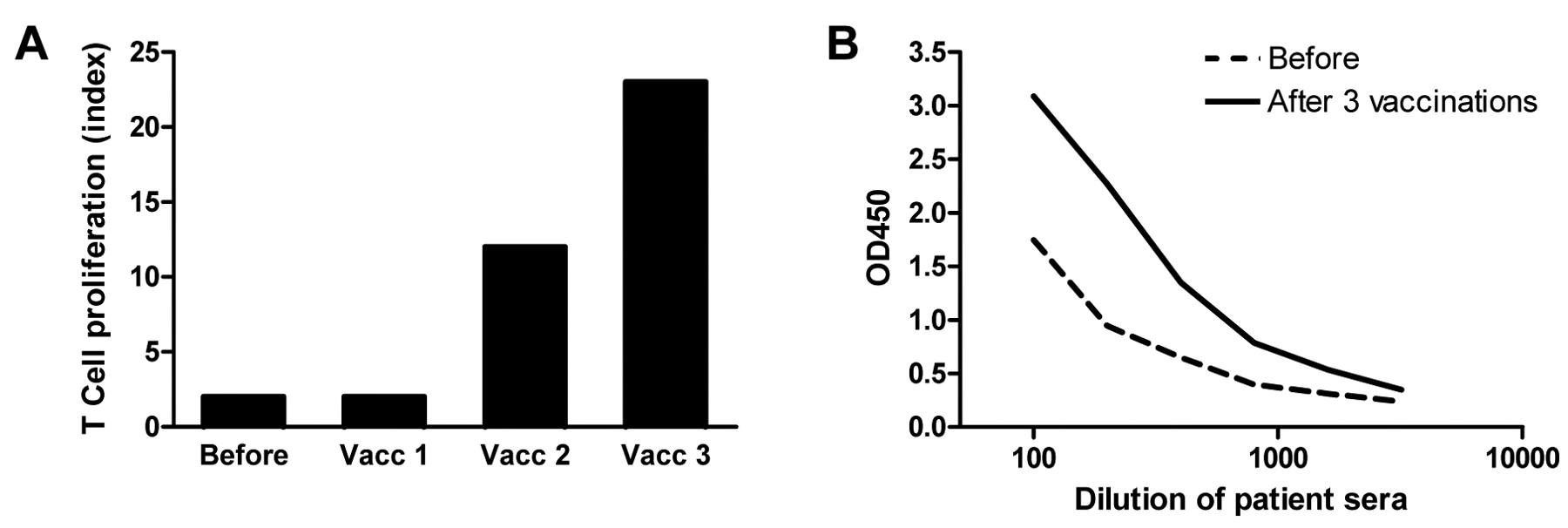
KLH responses. A: KLH-specific proliferation of PBMCs before and after one, two and three DC vaccinations of patient RNA-1. Proliferative response to KLH is given as proliferation index (proliferation +KLH/proliferation −KLH). B: Humoral responses against KLH in the serum of patient RNA-5. Serum was obtained before (dotted line) and after three vaccinations (solid line). Results of T-cell and humoral responses to KLH of all patients are shown in Table I.
DC vaccine characteristics. In all patients sufficient amounts of monocyte-derived mature DCs were obtained for three i.d. vaccinations (see Table I). However, the yield was not always sufficient for simultaneous i.v. injections. Patients vaccinated with CEA peptide-pulsed DCs received an average of 4×106 DCs i.v. and 5×106 DCs i.d. per vaccination during the first cycle. Patients vaccinated with RNA-electroporated DCs received an average of 11×106 DCs i.v. and 5×106 DCs i.d. (Table I). Final DC vaccine products met the criteria of a mature phenotype (3, 17), with high HLA-ABC, HLA-DR/DP, HLA-DQ, CD80, CD83, CD86 and CCR7 expression (Figure 1A). There was no difference in the maturation status (p=0.09 to p=0.88) or number of i.d. injected DCs. However, patients in the peptide group received fewer i.v. injected DCs (p<0.001). All patients had a proliferative peripheral blood CD4+ T-cell response against the control protein KLH (Table I), as expected for mature DC vaccine (17), indicating that the DCs were able to induce de novo T-cell responses.
On average, 66% of the DCs electroporated with mRNA expressed CEA protein 2 h after electroporation (range 44-89%; Figure 1B).
KLH-specific immune responses. To investigate whether peptide-loaded DCs and mRNA-electroporated DCs have similar capacity to activate the patient's immune system in general, humoral and cellular responses to the control protein KLH were measured in the peripheral blood of patients. PBMCs collected after each DC vaccination were analyzed for the presence of KLH-reactive T-cells in a proliferation assay. In both the peptide group and the RNA group, all patients tested showed a cellular response to KLH (Figure 2A). KLH-specific IgG antibodies were induced after vaccination in 8 out of 10 patients tested (80%) in the peptide group and in 2 out of 5 patients tested (40%) in the RNA group (Figure 2B). Thus, all patients developed a response to KLH, either cellular or humoral. However, more patients vaccinated with peptide-loaded DCs developed a humoral response to KLH. These results indicate that there may have been differences in the immunogenicity of DCs in both groups. Nevertheless, all patients had a proliferative T-cell response against KLH, indicating that the DCs were able to induce de novo T-cell responses in all patients.

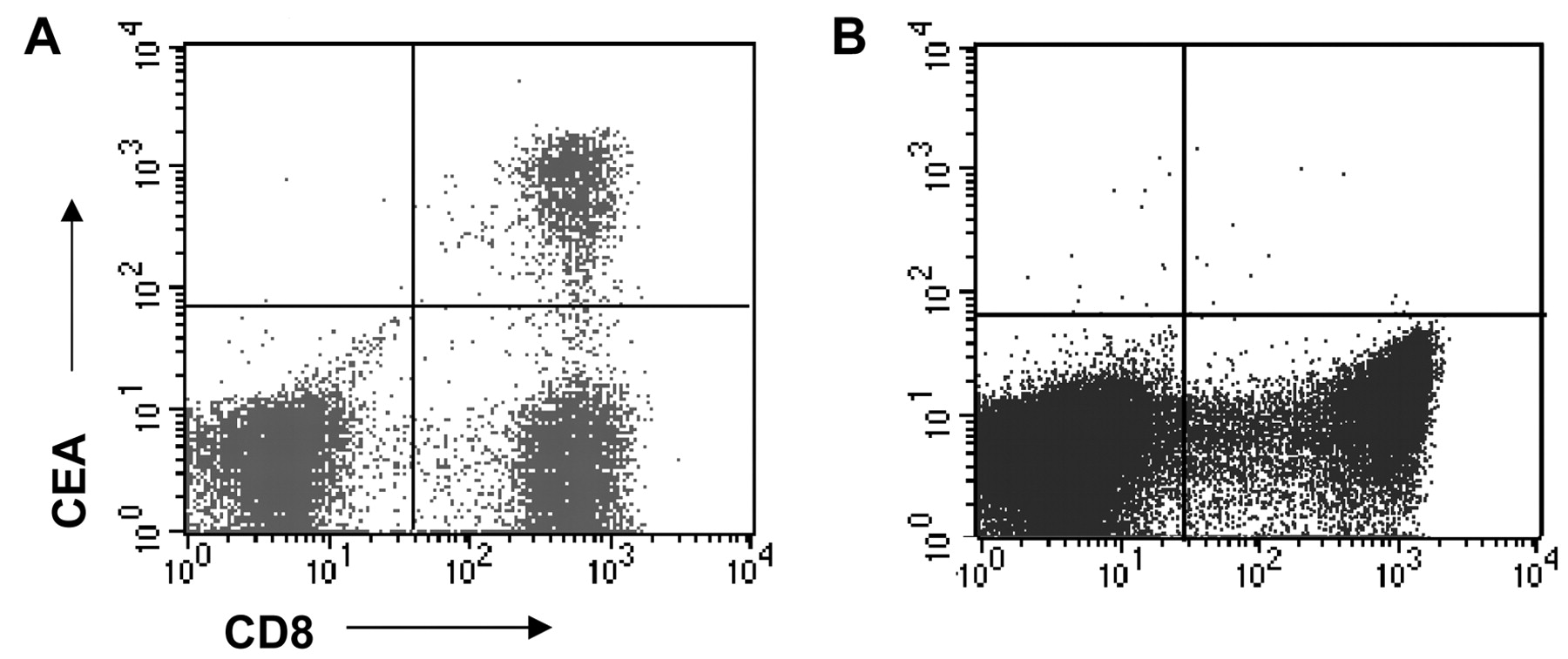
Tumor antigen-specific T-cell responses in post-treatment DTH. Tetramer analysis by flow cytometry of T-cells derived from a biopsy of a DTH reaction of patient pep-2 (A; CEA) and RNA-3 (B; DCs electroporated with CEA-encoding mRNA). Tetramer staining of 1 log above the negative population was considered positive. Tumor antigen-specific T-cell resonses of all patiens are shown in Table I.
Post-treatment DTH: CEA-specific T-cell immunity. To investigate the immune response against tumor peptides generated in vaccinated patients, DTH challenges were performed with mature DCs loaded with CEA peptide or CEA mRNA. Biopsies were taken from DTH induration sites, which were cultured with low amounts of IL-2 without the addition of antigen. In 8 out of 11 patients, CEA-specific DTH-infiltrated T-cells were detected by tetramer analysis after vaccination. We found that 0.3% to 98% of CD8+ T-cells were CEA-specific in DTH biopsies of patients vaccinated with HLA-A2 binding peptide-loaded DCs (Table I; partially previously reported in (13)). When these T-cells were co-cultured with CEA peptide-loaded T2 target cells, they were able to produce large amounts of interferon (IFN)γ and IL-2, but not IL-4 or IL-10 (Table I and (13)). An example of specific T-cells in a DTH site is shown in Figure 3A. In Table I, the presence or absence of CEA-specific T-cells at DTH sites is presented for each patient. In none of the patients vaccinated with mRNA-electroporated DCs was CEA-specific T-cell reactivity found in DTH biopsies (Figure 3B and Table I), despite the presence of skin induration in these patients (data not shown).
Discussion
Colorectal cancer is an immunogenic type of cancer. Tumor-associated antigens have been identified, tumor-specific T-cells have been isolated from patients and small proof-of-concept studies have shown that immunotherapeutic strategies can induce immunological and clinical responses (20-24). CEA is a tumor-associated antigen that is expressed by almost all colorectal tumors (25, 26). Hence, it is an attractive antigen to use in clinical immunization protocols. However, because CEA is also expressed in normal tissues, and it is also shed from the surface of tumor cells, a high threshold of tolerance must be overcome (27). Previously, it has been demonstrated that DCs loaded with CEA peptide can induce robust immune responses in colorectal cancer patients (13, 20, 28-31). Theoretically, using CEA mRNA instead of peptides to load the DCs may result in the induction of a much broader, more robust T-cell repertoire, since more epitopes are expressed and posttranslational modifications can occur.
For this reason, we compared CEA peptide-loaded DCs with CEA mRNA-electroporated DCs as vaccine in colorectal cancer patients. Previously, we have shown that with this technique, clinical-grade DCs can be efficiently transfected with tumor antigens, resulting in prolonged antigen-presentation and efficient T-cell activation (10). In all patients in the mRNA group, CEA expression was high upon electroporation of the DCs. The two DC vaccines were phenotypically identical. In addition, T-cell reactivity against the control protein KLH was comparable in the two groups after vaccination, indicating that the T-cell-stimulatory potential of the two DC vaccines were comparable. However, in 8 out of 11 patients who were vaccinated with peptide-pulsed DCs, we found CEA peptide-specific T-cell reactivity in DTH skin tests upon vaccination, whereas we could not detect CEA peptide-specificity in the 5 patients who were vaccinated with mRNA-electroporated DCs.
Monitoring immune responses in patients who are vaccinated with mRNA-transfected DCs poses a difficulty because the precise epitopes are not known. Therefore, the absence of tetramer-positive T-cells in the DTH tests should be interpreted with caution; it is possible that T-cells with specificity for other epitopes are in fact induced. However, in a previous melanoma study, using the same DC culture and electroporation protocol but different antigens (Gp100 and tyrosinase), we were able to demonstrate tetramer-specific T-cells in 8 out of 11 vaccinated patients (9). In addition, we also used mRNA-electroporated PBMCs as stimulator cells in co-culture with DTH-infiltrated T-cells upon vaccination for monitoring purposes. Nevertheless, we observed no T-cell specificity after vaccination in the mRNA group, which may depend on the antigen characteristics as well as the nominal epitope derived from the antigen.
In contrast to studies in melanoma (9), these results indicate that using CEA mRNA electroporation as a DC antigen loading strategy is not superior to peptide loading. The reasons for this difference may be twofold. Firstly, CEA is not processed through the MHC class II pathway whereas Gp100 is, unless it is coupled to a lysosomal targeting signal (32). Secondly, a different T-cell repertoire may be present in the vaccinated patients.
This study was not designed to allow meaningful conclusions on clinical efficacy. We did observe a transient increase in serum CEA upon vaccination in several patients, which may indicate a cytotoxic effect on CEA-expressing tumor cells (33).
In conclusion, in our study we could not find a benefit for CEA mRNA electroporation over peptide-pulsing as a method for antigen loading of DCs for vaccination in colorectal cancer patients.
Acknowledgements
This work was supported by grants from the Netherlands Organization for Scientific Research (920-03-250), and the TIL foundation. We kindly thank Jeanette Pots, Michel oldeNordkamp, Inge Boullart, Christel van Riel and Michelle Van Rossum for their assistance.
Footnotes
-
Authors' contributions
WJL treated the patients, performed the immunomonitoring and participated in study design and manuscript preparation. GS carried out the immunomonitoring and manuscript preparation. AdB, NMS and MvdR performed the DC culture and immunomonitoring. DHS performed and optimized the electroporation procedure. EHA and EJH included and treated the patients. CGF, GJA and JdV participated in the study conception and design and manuscript editing. CJAP participated in the study design, the treatment of the patients and manuscript editing. All authors read and approved the final manuscript.
-
Conflicts of Interest
The Authors declare that they have no competing interests.
- Received September 8, 2010.
- Revision received November 1, 2010.
- Accepted November 2, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A pilot study of the immunogenicity of a 9-peptide breast cancer vaccine plus poly-ICLC in early stage breast cancer
- Augmentation of Epithelial Resistance to Invading Bacteria by Using mRNA Transfections
- Immunotherapy of Autologous Tumor Lysate-loaded Dendritic Cell Vaccines by a Closed-flow Electroporation System for Solid Tumors
- Skin-Test Infiltrating Lymphocytes Early Predict Clinical Outcome of Dendritic Cell-Based Vaccination in Metastatic Melanoma
- Long-term Vaccine Therapy with Autologous Whole Tumor Cell-pulsed Dendritic Cells for a Patient with Recurrent Rectal Carcinoma