


Abstract
Background: To test whether intratumoral gene expression levels and germline polymorphisms predict clinical outcome in metastatic colorectal cancer (mCRC) patients treated with cetuximab and bevacizumab plus irinotecan (CBI) vs. cetuximab and bevacizumab (CB)(BOND2). Patients and Methods: Genomic DNA was extracted for genotyping from 65 patients (31: CBI arm and 34: CB arm). Thirty five patients had tissue samples available for the gene expression assay (18: CBI arm and 17: CB arm). Results: High intratumoral gene expression levels of EGFR, VEGFR2 and NRP1 were associated with longer overall survival (OS) in patients receiving combined monoclonal antibodies with or without irinotecan. FCGR3A V158F, CyclinD1 A870G and EGFR R497K polymorphisms are associated with clinical outcome in patients received combined cetuximab and bevacizumab. Conclusions: Intratumoral gene expression levels of EGFR, VEGFR2 and NRP as well as polymorphisms in FCGR3A, CyclinD1 and EGFR could predict clinical outcome in mCRC patients enrolled in BOND2, independent of KRAS mutation status.
- Metastatic colorectal cancer
- gene expression level
- polymorphisms
- clinical outcome
- cetuximab
- bevacizumab
- irinotecan
Colorectal cancer (CRC) represents the fourth most common cancer in the United States. In 2010, an estimated 142,570 new cases will be diagnosed and 51,370 deaths will occur (1). 5-Fluorouracil (5-FU)-based traditional chemotherapy has been used for the treatment of metastatic colorectal cancer (mCRC) for over 40 years. With the addition of other chemotherapeutic drugs such as irinotecan and oxaliplatin, the median survival of patients with advanced CRC has almost doubled from 12 months to 21 months (2). Recently, monoclonal antibodies that block either the epidermal growth factor receptor (EGFR) pathway or the vascular endothelial growth factor (VEGF) pathway have shown efficacy in the treatment of advanced CRC. Cetuximab is a human-murine chimeric IgG1 monoclonal antibody that targets the EGFR. It has shown antitumor activity both alone and in combination with irinotecan in irinotecan-refractory CRC (2, 3). Bevacizumab is a humanized anti-VEGF monoclonal antibody. It has been shown to improve clinical outcome when used in conjunction with irinotecan, fluorouracil and leucovorin chemotherapy, compared to chemotherapy alone (4).
The BOND-2 study was a randomized phase II trial in irinotecan-refractory mCRC patients and showed that when bevacizumab was added to cetuximab, or to cetuximab plus irinotecan, toxicities were as would have been expected from the individual agents and response rates and time to tumor progression appeared to compare favorably with historical controls (5). One challenging question for the management of mCRC is how to select patients who are most likely to respond and benefit from bevacizumab and cetuximab containing regimens, or, perhaps more importantly, how to select those patient who will not benefit, or in fact might suffer some detriment, from the treatment with a particular agent. Therefore, predictive and/or prognostic molecular markers could potentially maximize the clinical outcome and minimize toxicity as well as the patient's financial burden.
Demographic and clinical characteristics of patients by treatment arm in the subset pharmacogenetic study and the overall phase II trial population.
Several recent studies found that the K-RAS mutation status and the expression levels of epiregulin and amphiregulin are associated with the activity or inactivity of cetuximab (6-8). It was previously demonstrated that the gene expression levels of COX-2, EGFR, IL-8 and VEGF may be useful molecular markers of the clinical outcome in mCRC patients treated with single agent cetuximab (9). It was also shown that the gene polymorphisms involved in either EGFR pathway (Cyclin D1 or EGF) or in ADCC (FCGR2A/3A) are associated with the clinical outcome in a small group of mCRC patients treated with single agent cetuximab (10, 11). Graziano et al. showed in 110 advanced CRC patients treated with cetuximab plus irinotecan, that EGFR and EGF polymorphisms are associated with overall survival (OS) (12). There are no pharmacogenetic markers for bevacizumab in mCRC. However, a recent study by Schneider et al. found a significant association between VEGF genotype and median OS when using bevacizumab in metastatic breast cancer (13). In a previous study, there was no statistically significant association between the mutation of K-RAS, B-RAF or p53 and the response in a phase III trial with the addition of bevacizumab to first line irinotecan, 5-FU and leucovorin (IFL) in mCRC, neither were associated with progression-free survival (PFS) nor with OS (14). Preliminary data showed that the angiogenesis related gene polymorphisms may be associated with the clinical outcome in mCRC patients treated with first line 5-FU or capecitabine in combination with oxaliplatin and bevacizumab (FOLFOX/BV or XELOX/BV) (15).
Based on these studies, the present study examined the gene expression levels of 8 genes and a panel of 23 germline polymorphisms located in 21 candidate genes involved in the EGFR-related pathway (EGFR, EGF, COX-2, E-Cadherin, FCGR3A, FCGR2A/2B and Cyclin D1), the angiogenesis pathway (VEGF, IL-8, CXCR1, TGF-β, HIF1-α and NRP-1), the DNA repair pathway (ERCC1, XRCC1 and XPD) and the drug metabolism pathway (GSTP1, UGT1A1, ABCB1 and OATPC). These gene expression levels and germline polymorphisms were tested to predict the clinical outcome in mCRC patients enrolled in the BOND-2 trial.
Patients and Methods
Eligible subjects and treatment. Patient characteristics, recruitment and trial design have been described elsewhere (5). 83 patients with histopathologically confirmed mCRC, who received either cetuximab, bevacizumab and irinotecan (CBI arm, n=43) or cetuximab and bevacizumab alone (CB arm, n=40) were enrolled in the BOND-2 study. For the molecular correlate study, genomic DNA was extracted from peripheral white blood cells for genotyping from 65 patients. These included 31 patients from the CBI arm and 34 patients from the CB arm. For the gene expression assay, 35 patients had formalin-fixed paraffin-embedded samples available for microdissection of tumor DNA. Eighteen of these 35 patients were from the CBI arm and 17 were from the CB arm. Patients included in our molecular correlate analysis had similar demographic and pathologic characteristics and clinical outcome compared with the total patient population in the BOND-2 study (Table I). The investigations were conducted at the University of Southern California/Norris Comprehensive Cancer Center and were approved by the Institutional Review Board at the Keck School of Medicine, University of Southern California. All patients signed an informed consent for tissue and blood collection for the study of molecular correlates. Blood samples were collected before initiation of chemotherapy.
Gene expression assay and genotyping. Microdissection of tumor DNA, RNA isolation and cDNA synthesis and real time PCR quantification of mRNA expression levels of the targeted genes has been previously described (16). Briefly, paraffin-embedded tumor blocks were reviewed for quality and tumor content by a pathologist. 10 μm-thick sections were obtained from the identified areas with the highest tumor concentration. If the histology of the samples was homogeneous and contained more than 90% tissue of interest, the samples were dissected from the slides using a scalpel. All other sections were selectively laser capture microdissected according to the standard procedure. After cDNA was prepared by reverse transcription, quantitation of the candidate genes and of an internal reference gene (β-actin) was performed using a fluorescence-based real-time detection method (TaqMan) (Applied Biosystems, Forster city, CA, USA). Forward, reverse primers and probes were previously described (9, 16).
A whole blood sample was collected from each subject and genomic DNA was extracted from peripheral white blood cells. Single nucleotide polymorphism was tested using a polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) technique. Briefly, forward and reverse primers were used for PCR amplification, the PCR product was digested by restriction enzyme, and alleles were separated on 4% NuSieve ethidium bromide–stained agarose gel. The microsatellite repeat polymorphism in intron 1 of the EGFR gene and in the UGT1A1 gene was determined with a 5′-end 33P-γATP-labeled PCR protocol with a few modifications as previously described. Forward and reverse primers, restriction enzymes and annealing temperatures for the gene polymorphisms were described in previous publications (10, 11, 17, 18).
Statistical analysis. Time to tumor progression (TTP) was the primary endpoint in the clinical trial. Objective tumor response and OS were the secondary outcome variables. TTP was calculated as the time from the day of randomization until the first observation of disease progression or death from any cause. If a patient had not progressed or died, TTP was censored at the time of the last follow-up. OS time was calculated as the period from the day of randomization until death from any cause, or until the date of the last follow-up, at which point data were censored.
Gene expression values were expressed as ratios between 2 absolute measurements: that of the gene of interest and that of the internal reference gene, β-actin. The associations between gene expression levels and tumor response to therapy (partial response, stable disease and progressive disease) were evaluated by the Kruskal-Wallis test in the primary analysis. A classification and regression tree (CART) method, based on recursive partitioning (RP), was used to explore gene expression variables for identifying homogenous subgroups for survival (19). The RP analysis included all patients with tumor tissue specimen who were evaluable for measuring gene expression (n=35). Finally, to assess the associations between the expression level of each gene and TTP, the expression level was categorized into a low and a high value at optimal cutoff points. The maximal χ2 method of Miller, Siegmund and Halpern was used to determine which gene expression (optimal cutoff point) best segregated patients into poor- and good-prognosis subgroups (in terms of likelihood of tumor progression) (20, 21).
The association of each polymorphism with OS and TTP was analyzed individually using the Kaplan-Meier plots and the log-rank test. In the univariate analyses, the Pike estimate of relative risk (RR) with 95% confidence intervals (CI) was based on the log-rank test. The associations of each polymorphism with tumor response were summarized using contingency tables and the Fisher's exact test (Table II).
All tests of statistical significance were two-sided. The analyses were performed using the SAS statistical package version 9.0 (SAS Institute Inc. Cary, NC, USA) and Epilog Plus Version 1.0 (Epicenter Software, Pasadena, CA, USA).
Results
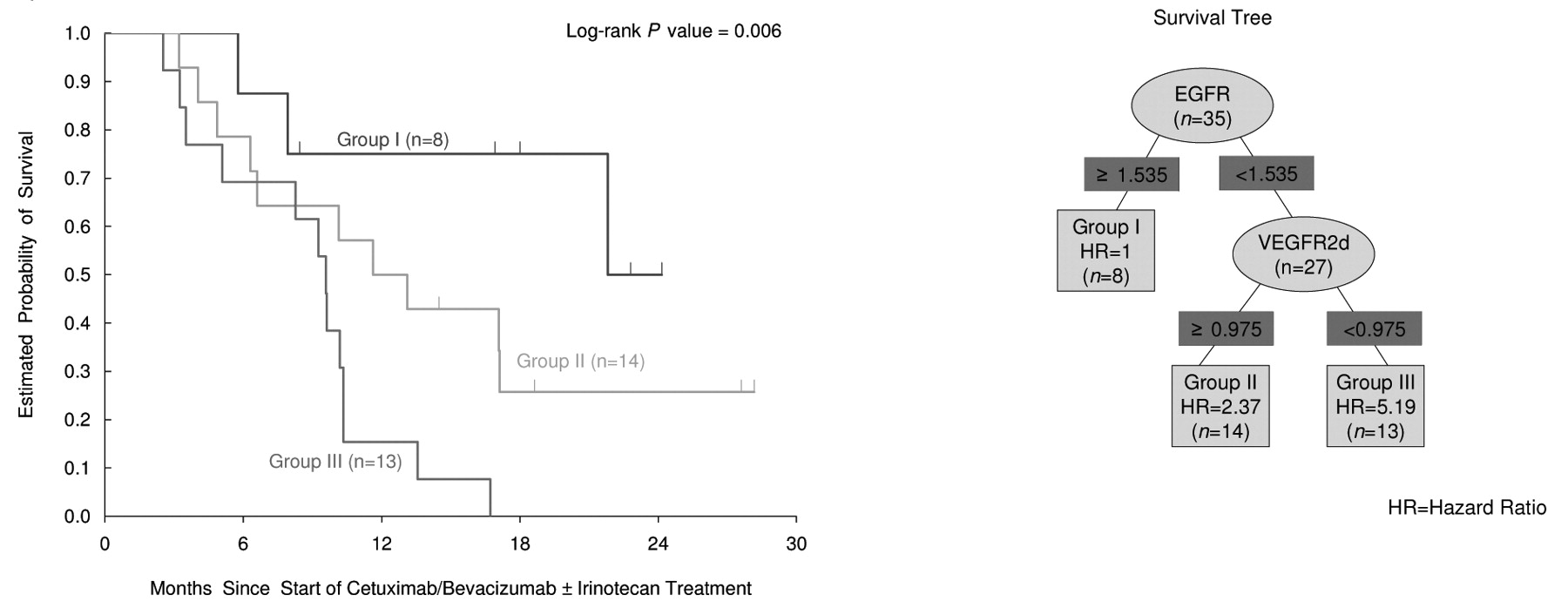
Gene expression levels and OS. Thirty five tumor tissues were available for gene expression assay, 18 from the CBI arm and 17 from the CB arm. Due to the small number of tissue samples, the gene expression levels and clinical outcome were analyzed in combination of both CBI and CB arms. There was no statistically significant difference of median gene expression levels between the 2 treatment arms (data not shown). CyclinD1 gene expression was quantifiable in 35 samples (100%), expression of ERCC1 and VEGF in 34 samples (97%) and expression of Il-8 and VEGFR2 in 32 samples (91%). Expression of NRP-1, EGFR and COX-2 were successful in 31 (89%) and 30 (86%) samples, respectively. The difference in the number of samples with quantifiable gene expression levels was due to the low or limited amount of cDNA/RNA generated from the microdissected paraffin-embedded tissues. High intratumoral mRNA levels of EGFR, VEGFR2 and NRP-1 were each significantly associated with OS in the univariate analysis (p≤0.05, Mann-Whitney U-test; Table III). Eight gene expression variables, treatment, K-RAS and p53 mutation status were considered for the RP analysis. The classification tree for overall survival is shown in Figure 1. Of the 9 gene expression variables evaluated, the RP analysis identified the EGFR expression level as the best single split with the best cutoff (1.535×10−3) between the better survival group (I) and the poor survival groups (II, III). The next best split in the lower EGFR group was the VEGFR2 expression level with the best cutoff (0.975×10−3) to further separate the poor survival group (II) (VEGF2≥0.975) and the worst survival group (III) (VEGF2<0.975). Among the patients with higher EGFR levels, no further categorizations could be identified. K-RAS mutation status was not chosen to predict survival in the RP analysis (data not shown). There were 8, 14 and 13 patients in the 3 terminal nodes I, II and III, respectively. Figure 1 shows the significant surviving difference between these 3 groups in the Kaplan-Meier survival curve.
(a) Baseline characteristics and clinical outcome in patients treated with the cetuximab/bevacizumab/irinotecan (CBI) arm. (b) Baseline characteristics and clinical outcome in patients treated with the cetuximab/bevacizumab (CB) arm.
Gene expression levels, TTP and tumor response. Expression levels of single genes analyzed in this study did not show any significant correlations with either tumor response or TTP in univariate analysis (Table III).
Germline polymorphisms and tumor response, TTP and OS. 65 patients had genomic DNA available for polymorphism analysis. Thirty-one of these patients were from the CBI arm and 34 were from the CB arm. The correlation between germline polymorphisms and tumor response, TTP and OS is listed in Tables IV and V.
Gene expression levels and gene polymorphisms are independent of K-RAS mutation status. K-RAS mutation status data were determined in a subset of patients treated at Memorial Sloan Kettering Cancer Center (MSKCC) (unpublished data). When this K-RAS mutation status was used as a parameter in the RP analysis of gene expression levels, it was not chosen to predict survival in the RP analysis (data not shown). Also, the significant gene polymorphism results were independent of K-RAS mutation status (data not shown).
Gene expression levels, time to tumor progression (TTP) and overall survival (OS) in univariate analysis.
Discussion
Targeted therapies, including the anti-EGFR antibody cetuximab and anti-VEGF bevacizumab, have shown efficacy in the treatment of mCRC. The BOND-2 study demonstrated that concurrent administration of these two monoclonal antibodies is feasible and the addition of bevacizumab to cetuximab plus irinotecan or cetuximab alone increases the anti-tumor activity compared with the historical control of cetuximab/irinotecan or cetuximab in bevacizumab-naïve patients. Identification of particular patients based on molecular markers, either in the tumor (tumor gene expression levels) or in the patient's genomic make up (germline polymorphisms), may select patients most likely to benefit either from individual antibody or from combined anti-EGFR and anti-VEGF therapies.
In this retrospective pharmacogenetic study, the gene expression levels and the germline polymorphisms involved in the EGFR and VEGF pathways were evaluated in patients enrolled in the BOND-2 study. High intratumoral gene expression levels of EGFR, VEGFR2 and NRP-1 were found to be associated with longer OS in patients receiving combined monoclonal antibody with or without irinotecan. Patients with high intratumoral EGFR gene expression levels had a median survival time of 21.8 (range, 9.6-28.2) months, compared to patients with low EGFR gene expression levels, whose median survival was 10.2 (range, 8.3-13.6) months (p=0.033). In the RP analysis, EGFR gene expression level was found to be the best single determinant to split patients into a better survival group (group I) or a poor survival group (groups II and III). The biological mechanism behind high intratumoral EGFR gene expression levels associated with better clinical outcome in mCRC patients treated with both cetuximab and bevacizumab is not clear. Previous studies examining EGFR expression by immunohistochemistry (IHC) have failed to show a significant association with clinical outcome in cetuximab-treated patients. This is likely due to the fact that IHC is a semiquantitative and subjective method which is limited by the sensitivity of the monoclonal antibody and the tissue handling (3). Several studies did demonstrate that mCRC patients with a high EGFR copy number, measured by fluorescence in situ hybridization (FISH), had an increased likelihood to respond to cetuximab therapy. Cappuzzo et al. found that patients with EGFR genomic gain (EGFR copy number >2.92 by FISH) were associated with response to cetuximab (19). Jimeno et al. tested whether global activation of the EGFR pathway is predictive of EGFR inhibitor (erlotinib and cetuximab) efficacy in pancreatic cancer xenografts. Their data showed that EGFR pathway genes are highly expressed in tumors sensitive to EGFR inhibitors compared with tumors resistant to EGFR inhibitors (22). Another in vitro study by Fujimoto et al. also demonstrated the positive relationship between the high expression of EGFR family members (EGFR, ErB2 and ErB3) and the sensitivity to EGFR inhibitor gefitinib in three different models, namely human lung adenocarcinoma cell line, mouse model and tumor biopsies from lung cancer patients (23).

Overall survival (OS) of different gene expression group by recursive partitioning (RP) analysis.
The results of the present study also showed that high intratumoral gene expression levels of two VEGF receptors, VEGFR2 and NRP-1, are associated with a better OS. Patients with higher VEGFR2 levels had a median survival time of 13.1 (range, 9.6-21.8) months versus patients with lower VEGFR2, who had a median survival time of 9.6 (range, 5.1-10.2) months (p=0.049). Patients with higher NRP-1 gene expression levels had a median survival time of 11.6 (range, 9.6-21.8) months compared with patients with lower NRP-1 expression, who survived for only 5.1 months (p=0.012). In the RP analysis, VEGFR2 gene expression was the second best determinant after EGFR expression to further split patients into a modest survival group (group II) and a worse survival group (group III). VEGFR2 and NRP1 are co-receptors for VEGF, and previous studies have shown that these two receptors are expressed in gastrointestinal tumor cells. The data of the present study are consistent with an in vitro study by Calvani et al., which demonstrated that colon cancer cells differentially express a functional VEGF/VEGFR2/HIF-1α autocrine loop that mediates survival under hypoxic conditions (24). More importantly, inhibition of VEGF by bevacizumab induced cancer cell apoptosis only in a VEGF-sensitive cell line (HCT116), which expresses high levels of VEGFR2 and NRP-1, but not in a VEGF–resistant cell line (HT29), which lacks a functional VEGF/VEGFR2/NRP-1 signaling pathway (24). Wedem et al. also suggested that blockade of VEGF by bevacizumab inhibits the activation of VEGFR2 in tumor cells and induces tumor apoptosis (25).
Although intratumoral gene expression levels are potentially valuable molecular markers, the patient's inherited genetic make up, namely the germline polymorphisms, may also play an important role in determining the patient's clinical outcome when treated with a combined anti-EGFR and anti-VEGF therapy. Cetuximab is a chimeric- and bevacizumab is a humanized-IgG1 monoclonal antibody. Previous studies found that cetuximab may exert its antitumor effect through antibody-dependent cell-mediated cytotoxicity (ADCC) (26). Previously, two functional polymorphisms in FCGR2A and FCGR3A were associated with clinical outcome in mCRC patients treated with single-agent cetuximab (11). In the current study, FCGR3A V158F polymorphism was significantly associated with the patient's response to combined cetuximab and bevacizumab therapy (CB arm). A total of 56% of patients with F/F genotype responded to cetuximab plus bevacizumab treatment, compared to 25% heterozygous F/V and 8% homozygous V/V genotype (p=0.05). Similar data were also reported to HER-2/neu-positive metastatic breast cancer patients treated with trastuzumab-based therapy and in follicular lymphoma patients treated with rituximab (27, 28). The results of the present study also confirmed that Cyclin D1 A870G polymorphism and EGFR R497K polymorphism are associated with either TTP or OS in the CBI arm, which was consistent with previous findings (10, 18).
(a) Significant associations between germline polymorphisms and tumor response in univariate analysis. (b) Significant associations between germline polymorphisms and time to tumor progression (TTP) in univariate analysis.
The present pharmacogenetic study is a small, retrospective, hypothesis generating study. For this reason, these preliminary findings have limitations and results should be interpreted cautiously. The first limitation is the relatively small number of patients enrolled in the BOND-2 study (n=35 for gene expression; n=65 for germline polymorphisms). Secondly, the study examined 8 gene expression levels and 23 polymorphisms in 21 genes associated with the EGFR and VEGF pathways. These candidate genes were selected based on their functional role in the EGFR and VEGF pathways or based on relevant previous data and publications (9). Due to the limited number of patients, a multivariate analysis was not feasible. Therefore, RP analysis was used as an internal validation analysis to reduce the possible bias by univariate analysis.
Significant associations between germline polymorphisms and overall survival (OS) in univariate analysis.
Notwithstanding the aforementioned limitations, the present study suggested that high intratumoral EGFR, VEGFR2 and NRP-1 gene expression levels are associated with longer OS in mCRC patients treated with combined cetuximab and bevacizumab. Polymorphisms involved in the EGFR and VEGF pathways may predict clinical outcome in this patient population. In the post genomic era, findings such as these will not only help identify patients who are most likely to benefit from targeted therapies, but they will also be critical in reducing potential chemotherapy toxicity and minimize the patient's financial burden. Larger, prospective clinical trials such as the CALGB 80405 or the CAIRO-2 trial are warranted to validate these preliminary findings.
Acknowledgements
This work was funded by the U.S. National Institutes of Health grant 5 P30CA14089-27l, the San Pedro Guild Research Fund and DHONT Foundation and grants from Genentech, Inc. and Imclone systems, Inc.
Footnotes
-
↵* Both authors contributed equally to this paper.
-
Conflicts of Interest
Dr. Heinz-Josef Lenz: Consultant or advisory role: Genentech, BMS; Research funding: Genentech; Dr. Leonard Saltz: Consultant or advisory role: Genentech, BMS; Research funding: Genentech; Kathleen D Danenberg is the CEO and owns stock in Response Genetics, Inc. All other Authors state that they have no conflicts of interest.
- Received June 29, 2010.
- Revision received July 12, 2010.
- Accepted July 26, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
Jump to section
Related Articles
Cited By...
- Pharmacogenomics, biomarker network and allele frequencies in colorectal cancer
- Fc-{gamma} Receptor Polymorphisms, Cetuximab Therapy, and Survival in the NCIC CTG CO.17 Trial of Colorectal Cancer
- Fc{gamma}RIIa and Fc{gamma}RIIIa Polymorphisms and Cetuximab Benefit in the Microscopic Disease
- Vascular Endothelial Growth Factor Pathway Polymorphisms as Prognostic and Pharmacogenetic Factors in Cancer: A Systematic Review and Meta-analysis
- Anti-EGFR Antibody Cetuximab Enhances the Cytolytic Activity of Natural Killer Cells toward Osteosarcoma
- Biomarker Use in Colorectal Cancer Therapy