


Abstract
Aim: The effect of combining sodium butyrate (NaB), a histone deacetylase inhibitor, and 7-hydroxy-staurosporine (UCN-01) on cytotoxicity in human cervical carcinoma cells was evaluated. Materials and Methods: HeLa and CaSki cells were treated using NaB alone or in combination with staurosporine (STS) or its analog UCN-01. Cytotoxicity was determined by flow cytometry and morphological assays. Apoptotic pathways were characterized by Western blotting and immunostaining. CaSki cells were also xenografted into nude mice to assess the in vivo effects of NaB/UCN-01 combination. Results: Treatment with NaB and STS or UCN-01 resulted in enhanced apoptosis of cancer cells. Apoptosis involved mitochondrial pathways and overexpression of p53 and p73. In concordance, co-treatment modulated some p53/p73 downstream targets such as p21, BAX, BCL-2 and BCL-XL, leading to increased caspase-3 and poly(ADP-ribose) polymerase cleavage. In vivo, NaB/UCN-01 combination exerted a substantial tumour growth suppression effect compared with single treatment. Conclusion: UCN-01 was shown to be a potentiator of NaB therapy for cervical cancer cells.
Cervical cancer is the second most common cancer among women worldwide. A key factor in cervical carcinogenesis is persistent infection by high-risk human papillomaviruses (HPV), especially by HPV16 and 18, associated with 70% of cervical tumours (1). Although recent clinical trials have shown that HPV16/18 virus-like particles are effective in prevention of HPV infection and cervical dysplasia, novel therapeutic strategies for established cervical cancer with minimal deleterious effects are needed to complement or replace existing therapy. Since 1999, cisplatin has been the standard chemotherapeutic agent to treat advanced, recurrent and metastatic squamous cell carcinoma of the uterine cervix. However progression-free and overall survival are still limited, even when cisplatin-based chemotherapy is administered during radiotherapy (2). During recent years, significant improvements in understanding altered molecular events in tumour cells have led to the discovery of new targets and agents for clinical testing (3). The implication of high-risk HPV in cervical cancer is due to the production of viral E6 and E7 oncogenes that are necessary and sufficient to acquire and maintain the malignant phenotype (4). These viral oncoproteins constitutively expressed after HPV DNA integration into the host genome can target histone acetyl transferase (HAT) and histone deacetylase (HDAC) enzymes. Indeed, HPV16 E6 is capable of abrogating the co-stimulatory function of CREB-binding protein (CBP) and p300 which normally possess an intrinsic HAT activity. As for E7 oncoprotein, it can directly bind to the HDAC complex (5). This property presumably allows E7 to modulate cellular genes with subsequent outgrowth of premalignant cells. E7 can also relieve the repressive effect of pRb and HDAC1 on the cyclin E promoter thereby promoting unscheduled cell cycle progression (6).
Recently, considerable attention has focused on the development of HDAC inhibitors (HDACi) as potential anticancer agents for the treatment of solid malignancies (7) and many HDACi have entered clinical trials. These drugs regulate gene transcription through chromatin remodelling (8) and modulate acetylation status of a series of non-histone proteins (9). Numerous studies reported that HDACi induce growth arrest, differentiation, activation of extrinsic and/or intrinsic apoptotic pathways, autophagic cell death, reactive oxygen species-induced cell death, mitotic catastrophe cell death and senescence in various transformed cells (7). As for the intrinsic apoptotic pathway activation, HDACi up-regulate proapoptotic proteins such as BIM, BAX or BAK, but the mechanism is, however, not well understood; they also down-regulate anti-apoptotic proteins such as BCL-2 and BCL-XL in a cell context-dependent manner (10-12). Additionally, HDACi can induce BID cleavage followed by mitochondrial disruption (13). Lastly, it has been demonstrated that they suppress transcription of X-linked inhibitor of apoptosis protein (XIAP) (11, 12) and favour survivin degradation (12).
HDACi belong to a heterogeneous class of compounds. Early efforts centered on sodium butyrate (NaB), a short-chain fatty acid, and trichostatin A (TSA), a hydroxyaminic acid (14), which exert anti-proliferative and pro-apoptotic effects in many cell lines including cervical cancer cells (15). Both compounds increase cyclin-dependent kinase (CDK) inhibitors (CKI) p21WAF1/CIP1 and p27KIP1 with a concomitant loss of CDK2 activity, which leads to growth arrest in G1 phase of the cell cycle (15), despite ongoing HPV transcription (16). HDAC inhibition also leads to a time-dependent degradation of pRb, p107 and p130, releasing E2F-1 transcription factor. The presence of free E2F following initial G1 arrest and concomitant up-regulation of CKIs results in a ‘conflicting growth situation’, which finally leads the cells to undergo apoptosis by inducing the E2F-responsive proapoptotic α- and β-isoforms of p73 acting through the mitochondrial pathway (17).
Moreover, HDACi have shown synergistic, and additive anti-tumoural effects with a wide range of chemotherapeutic drugs (7). In this context, the ability of kinase inhibitors to potentiate the lethal effects of HDACi has been previously reported (18-21). However, synergism between NaB and staurosporine (STS), or its analog 7-hydroxy-staurosporine (UCN-01), has not been extensively evaluated except in Jurkat cells and colorectal cancer cells (22).
STS is a lead compound among broad-spectrum kinase inhibitors (23-24). It has been widely used as an inducer of apoptosis in cellular studies (25). In our laboratory, we have demonstrated that STS induces apoptosis in wild-type p53 cervical carcinoma cell lines (HeLa and CaSki) to a higher extent compared with a mutant p53 cell line (C-33A) (26-28). We also reported that STS leads to inhibition of E6 and E7 oncogenes and mouse double minute 2 (MDM2) expression, increased levels of p53, p21WAF1/CIP1 and BAX and decreased levels of BCL-2 and BCL-XL (29). Because wild-type p53 and mutated p53 were transiently located to the mitochondria, we have suggested that STS-induced apoptosis is p53 dependent (27, 28). Moreover, we recently described that STS engages the intrinsic mitochondrial apoptotic pathway via caspase-8 or caspase-9 signalling cascades and via caspase-independent pathway (30).
As for UCN-01, it has been demonstrated to be more specific than STS, inhibiting conventional protein kinase C (PKC) isoenzymes more potently than novel and atypical ones and inhibiting better the membrane-derived PKC than the cytosolic one (31). UCN-01 causes accumulation in G1 phase in human epidermoid carcinoma cells (32), while it is a potent abrogator of G2 checkpoint control in cancer cells with disrupted p53 function (33). Other reports indicate that breast carcinoma and malignant glioma cells are susceptible to UCN-01-induced apoptosis (31, 34). This compound also exerts antitumour activity against different human tumour xenografts grown in nude mice (35, 36).
In view of the documented activities of NaB and STS, and its analog UCN-01, in malignancies, the possibility arises that combined treatment with these agents might result in enhanced antitumour activity. The aim of our study was to determine whether NaB together with UCN-01 exerts antiproliferative and pro-apoptotic activity in HPV-positive cancer cells and inhibits tumour growth in xenografted nude mice.
Materials and Methods
Cell lines. Two human cell lines, obtained from the American Type Culture Collection (ATCC; Rockville, MD, USA) were used in this study. HeLa and CaSki cells are HPV-positive cells derived from human cervical carcinomas with wild-type p53 (p53wt). HeLa and CaSki cells were respectively cultured in Eagle's minimal essential medium (EMEM; Bio Whittaker Europe, Verviers, France) and RPMI (Bio Whittaker) supplemented with 10% (v/v) foetal bovine serum (FBS) (Sigma, St Louis, MO, USA), 2 mM L-glutamine (Bio Whittaker), 100 units/ml penicillin and 10 mg/ml streptomycin. Cells were incubated at 37°C under a humidified atmosphere of 95% air and 5% CO2 (v/v). They were routinely monitored and found to be free of mycoplasm infection.
Treatments. Prior to each treatment, cells were plated either onto 6-well plates with 5×104 cells per well or onto 75 cm2 flasks with 8×105 cells per flask, and grown until 50% of confluence. Growth medium was removed and replaced by treatment medium. To induce apoptosis, the following drugs were used: (i) NaB (Sigma Aldrich, Saint Quentin Fallavier, France) was dissolved in culture medium for in vitro studies. Working concentrations were between 0.6 mM and 100 mM. For in vivo studies, NaB was dissolved in sterile water. (ii) UCN-01 (Sigma Aldrich) was reconstituted in DMSO as a 2 mg/ml stock solution, which was further diluted to the working concentration (50-1000 nM) in culture media. The final concentration of DMSO did not exceed 0.5%. For in vivo studies, UCN-01 was diluted in sterile water containing 2% sodium citrate (pH 3.5). (iii) STS (Sigma Aldrich) was also reconstituted in DMSO as a 200 μM stock solution with working concentrations between 1 nM and 200 nM.
Cell cycle analysis and assessment of cell death. At the indicated times, cells were washed twice with PBS, harvested by trypsinization (Bio Whittaker), washed again twice with PBS and centrifuged at 300×g for 10 min. For cell cycle and sub-G1 DNA content analysis, a batch of cells was resuspended and fixed overnight in 70% (v/v) cold ethanol. Fixed cells were then washed twice with cold PBS before the addition of 1 mg/ml RNaseA DNase-free plus 10 μg/ml propidium iodide. After 15 min at room temperature (RT), cells were analysed by flow cytometry on a FACScan Epics Altra flow cytometer (Beckman Coulter, Fullerton, CA, USA). Twenty thousand events were collected for each sample. Cell cycle analyses were performed on a viable gated cell population using Wincycle software (Phoenix Flow Systems, San Diego, CA, USA) and the percentage of cells with sub-G1 DNA was calculated using Expo-32 software (Beckman Coulter).
For measurement of mitochondrial membrane potential (ΔΨm), another batch of cells was resuspended in 200 nM MitoTrackerRed CMXRos (Molecular Probes Europe, Invitrogen, Paisley, UK) diluted in the appropriate culture medium according to the cell type and incubated at 37°C for 1 h. Cells were then washed twice with PBS, centrifuged and resuspended in 400 μl PBS for consecutive flow cytometric analysis on Cell Quest (FACSort, Becton Dickinson). Measurement of the mean fluorescence intensity was used to assess the variations in ΔΨm. The percentage of cells with depolarized mitochondria was furthermore determined. Twenty thousand events were collected for each sample.
Immunostaining of intracellular proteins. For measurement of intracellular expression level of BCL-2, BCL-XL and BAX proteins, the following antibodies from Beckman Coulter were used: purified monoclonal antibody (mAb) anti-BCL-2 (clone 83-8B), fluorescein-conjugated anti-BCL-XL (clone 7B2.5) and purified mAb anti-BAX (clone 4F11). One to two hundred thousand cells per sample were fixed and permeabilized using IntraPrep™ Kit according to the manufacturer's instructions (Beckman Coulter) and incubated either with the relevant antibody or its isotypic control for 20 min at RT. Cell samples were washed in PBS and incubated for 1 h at RT with a fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-coupled secondary antibody when it was required. After an ultimate wash, cells were immediately analyzed on Cell Lab Quanta flow cytometer (Beckman Coulter) using Cell Lab Quanta SC software.
For measurement of intracellular cleaved caspase-3, 1×105 to 2×105 cells per sample were fixed by incubation for 10 min at 37°C with 1.5% formaldehyde, refreshed for 1 min on ice and permeabilized for 10 min on ice with cold 90% methanol. Cells were then washed twice in cold PBS and once in incubation buffer (PBS containing 5 mg/ml bovine serum albumin). After 10 min at RT in 90 μl of incubation buffer, cells were stained for 1 h (RT in the dark) with 7.5 μl of (Asp175)-Alexa fluor-anti-human caspase-3 antibody. Finally, samples were washed in buffer and immediately analyzed on Cell Lab Quanta flow cytometer.
Immunoblotting. Briefly, after indicated periods of treatment, cells were collected in RIPA lysis buffer containing anti-proteases and proteins were extracted from whole cells by cell disrupting. Protein concentration was quantified using Biorad Protein Assay® (BioRad, Hercules, CA, USA) according to the manufacturer's instructions. Twenty μg of proteins were boiled in Laemmli's buffer (v/v) (Biorad) supplemented with 5% β-mercaptoethanol and run on SDS polyacrylamide gel. Proteins were then electrotransferred onto Hybond™ membranes (Amersham, Saclay, France), probed for 1 h with appropriate primary antibodies (see below). After rinsing, immune complexes were revealed with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibodies for 1 h (Pharmingen, San Diego, CA, USA). The reaction was visualized by autoradiography using ECL reagents (GE Healthcare, Diegem, Belgium) according to the manufacturer's instructions.
Monoclonal primary antibodies were purchased from PharMingen: p53 (1:500, clone DO-7); TA p73 (1:500, clone GC-15), p21WAF1/CIP1 (1:250, clone 6B6); PARP-1 (1:1000, clone 4C10-5). The rabbit polyclonal anti-procaspase-3 and cleaved fragment (1:500, clone AAP-113) was purchased from Stressgen (Ann Arbor, MI, USA). Equal protein loading and transfer was routinely monitored by incubating membranes with a beta-actin specific antibody (1:10,000; clone AC-15; Sigma).
Animals, tumour xenografts and treatment. All animal studies were carried out under an appropriate accreditation and all experiments were performed in compliance with the French Ministry of Agriculture regulations for animal experimentation. Female athymic (nu/nu) nude mice, 4-8 weeks old and weighing 18-21 g, used in the study were purchased from Charles River (L'Arbresle, France), housed in appropriate sterile filter-capped cages and allowed feed and water ad libitum (Institut Fédératif de Recherche 133). All handling and transplantation procedures were conducted under a laminar-flow biosafety hood. CaSki cells were used to induce xenografts in athymic mice as described previously (37). Briefly, exponentially growing cells were harvested, washed, resuspended in PBS 5% heat-inactivated FBS (Sigma), then 3.5-5×106 viable cells were grafted subcutaneously into the flank of mice. Animals were then monitored twice a week for weight and tumour formation. Drug treatment started when the tumours reached a volume between 0.05 and 0.2 cm3. A first series of experiments consisted of dose–response studies (200 and 800 mg/kg for NaB and 1.875 and 7.5 mg/kg for UCN-01). According to the results, animals were then randomized into 4 groups of 6 mice. The first group was the control group receiving only the vehicle, the second group was treated with the sub-optimal dose of NaB, the third group was treated with the sub-optimal dose of UCN-01 and the fourth group was treated with a combination of both drugs. Compounds were given by intraperitoneal injection, for 5 consecutive days per week, for 2 consecutive weeks. At the end of experiments, animals were ethically sacrificed for retrieval of tissue, which was fixed in 4% buffered formalin and paraffin-embedded. Individual tumour volumes were estimated as described previously (37) by the formula: tumour volume (cm3)=½×W2×L (where W is the width and L is the length of the tumour).
Histology and in situ hybridization analysis of tumour xenografts. The morphology of xenografts was studied using 5 μm sections and standard protocols. Immunohistochemical staining to evaluate proliferating cells was carried out using biotinylated primary antibody anti-Ki-67 (MIB-1, 1:150; DakoCytomation, Glostrup, Denmark). In situ hybridization was performed using specific biotinylated HPV16/18 DNA probe as we described previously in detail (38). CaSki cells served as positive control. Slides were examined and pictures were taken using a Zeiss Axioskop 40 photomicroscope.
Statistical analysis. Data are expressed as mean±SD of three independent experiments. For in vitro experiments, all comparisons were made with two-tailed unpaired Student's t-test. The in vivo therapeutic efficacy of NaB and UCN-01 was assessed by evaluating the tumour volume development over time with two-way ANOVA test. Differences between control and treated mice at each post-graft time were determined by Student's t-test. Values of p<0.05 were considered significant.

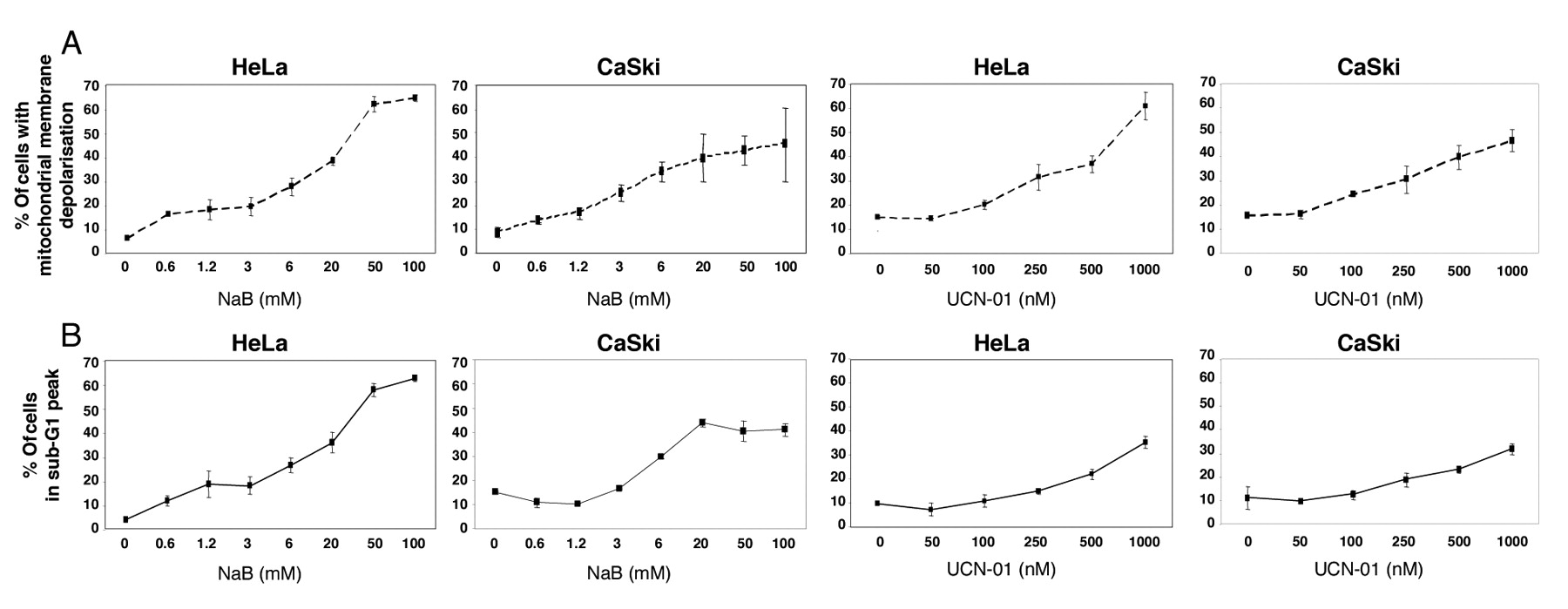
NaB and UCN-01 induce apoptosis in human cervical carcinoma cells. HeLa and CaSki cells were treated for 48 h with increasing concentrations of NaB, or for 24 h with increasing concentrations of UCN-01, as described in the Materials and Methods. A: Following treatment with NaB or UCN-01, mitochondrial membrane depolarization was analysed by flow cytometry using MitoTracker Red dye. B: Sub-G1 DNA levels were assessed following cell incubation with propidium iodide. All results represent the mean±s.d of 3 determinations. They are representative of 3 independent experiments.
Results
Effects of NaB and UCN-01 on cell cycle traverse and apoptosis of cervical-derived cancer cell lines. In order to test the effects of NaB and UCN-01, HeLa and CaSki cells were exposed to increasing concentrations of these two molecules. In accordance with the literature, our preliminary time-course experiments revealed that 48 h and 24 h were optimal periods for cell exposure to NaB and UCN-01 respectively (17, 26). In both cell lines, NaB induced a dose-dependent increase of the G1 fraction (e.g. from 60% in untreated CaSki cells to 80% in cells treated with 100 mM NaB), whereas it reduced the S phase fraction (e.g. from 26% to 9% for CaSki cells). NaB also increased the proportion of cells with mitochondrial ΔΨm collapse (Figure 1A) and fragmented DNA (Figure 1B). These data are in agreement with those found by Finzer et al. (15, 17).
We achieved parallel cultures of HeLa and CaSki cells incubated with UCN-01, a staurosporine analog. Treatment with this compound resulted in an increase of the G2/M fraction only at the highest doses tested (e.g. from 13% in untreated CaSki cells to 32% in cells treated with 1 μM UCN-01). It also induced loss of ΔΨm (Figure 1A) and apoptosis in a dose-dependent manner (Figure 1B), but less efficiently than did STS (data not shown). These findings are in agreement with our previous observations (26, 27).
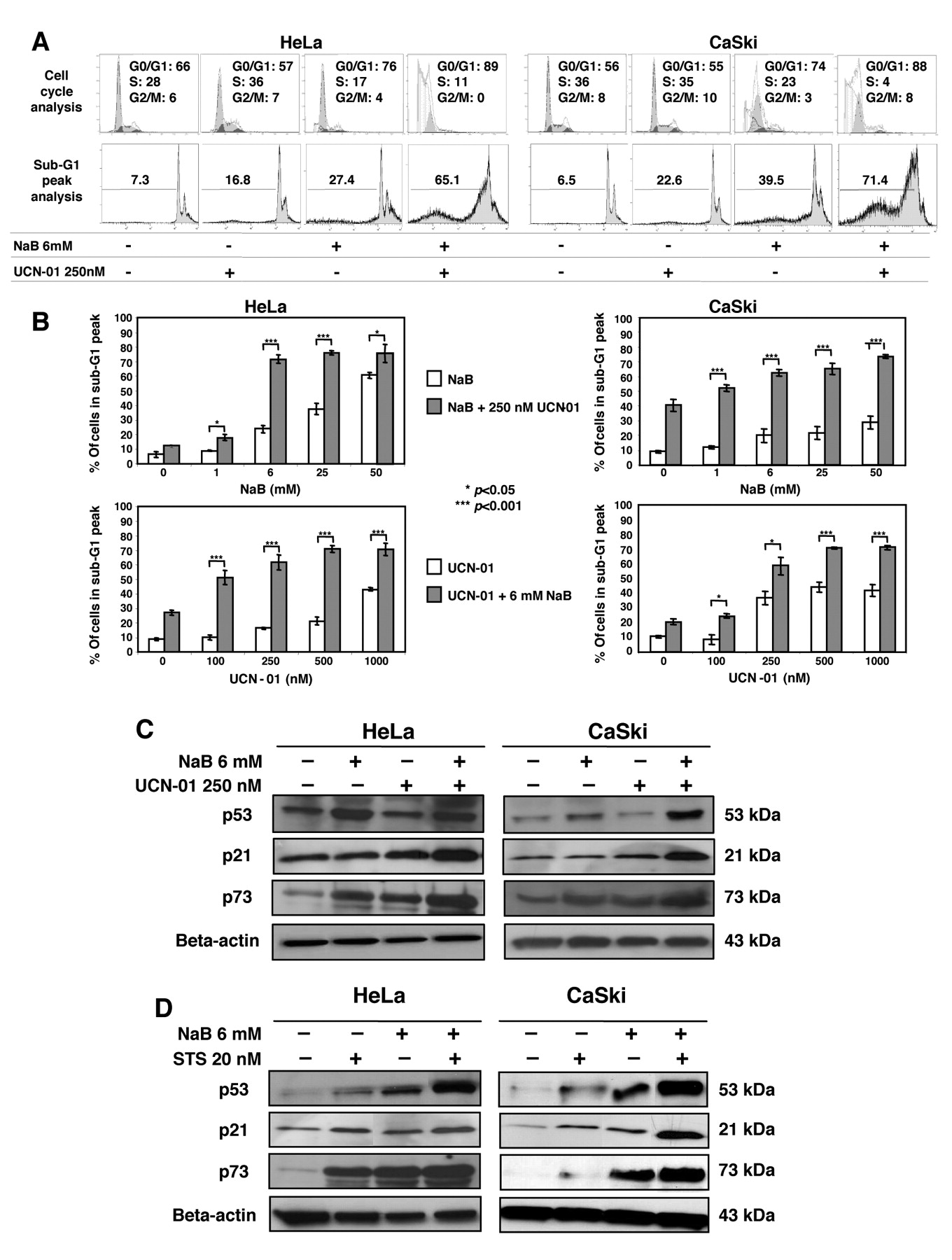
Combined exposure to NaB and UCN-01 or STS results in increased apoptosis in HeLa and CaSki cells. To investigate apoptosis induction after co-treatment with NaB and UCN-01 in HeLa and CaSki cells, several schedules were investigated. On the basis of our preliminary findings (not shown), studies focused on the effects of pretreatment with NaB for 24 h followed by UCN-01 for an additional 24 h. Thus, cells were either pretreated with increasing doses of NaB and then with a fixed dose of UCN-01 or pretreated with a fixed dose of NaB and then increasing doses of UCN-01. The fixed doses used (6 mM NaB and 250 nM UCN-01) were chosen based upon our dose–response studies (Figure 1), corresponding to a 20-30% cytotoxic effect in monotreatment. Figure 2A is representative of cell cycle and sub-G1 peak cytometric analyses, showing that exposure of cells to combinatory treatment significantly increased the G0/G1 fraction and reduced the S-phase fraction. Combination of 250 nM UCN-01 with increasing concentrations of NaB elicited greater DNA fragmentation in both cell lines compared with NaB alone (Figure 2B, upper panels). Similar results were obtained when cells were exposed to a fixed dose of 6 mM NaB with increasing doses of UCN-01 (Figure 2B, lower panels). In both experiments, 250 nM UCN-01 in conjunction with 6 mM NaB, which were modestly toxic by themselves, resulted in a significant increase in apoptosis after 48 h. In separate studies, comparable changes were noted in HeLa and CaSki cells exposed to 250 nM NaB and 20 nM STS (data not shown).
Potentiation of apoptosis in HeLa and CaSki cells exposed to NaB and UCN-01 or STS is associated with modulation of cell cycle regulators and BCL-2 family members. To further investigate the effects of NaB and UCN-01 or STS, we performed time-course experiments in which HeLa and CaSki cells were treated for 12 to 48 h with 6 mM NaB, or for 2 to 24 h with 250 nM UCN-01 or 20 nM STS. For the longer times, we demonstrated that 6 mM NaB induced p73 as well as p53 expression. We also found that STS, and to a lesser extent UCN-01, induced p53 and p73 expression (Figure 2C-D).

NaB and UCN-01 combinatory treatment induces G1 phase accumulation and enhances apoptosis in HeLa and CaSki cells through induction of cell cycle regulators. A: Cells were treated for 48 h with 6 mM NaB, or for 24 h with 250 nM UCN-01, or with a combination of both drugs. Flow cytometric analyses show cell cycle distribution in the G1, S and G2/M phases (upper panel) and sub-G1 DNA content after staining with PI (lower panel). Data are representative of multiple independent experiments. B: Histograms show the percentage of apoptotic cells after co-treatment with NaB and UCN-01, either with a fixed concentration of 250 nM UCN-01 plus increasing concentrations of NaB (upper panels), or with a fixed concentration of 6 mM NaB plus increasing concentrations of UCN-01 (lower panels). Each bar represents the mean±s.d of 3 determinations. Similar results were obtained from 3 independent experiments. *P<0.05, ***P<0.001. C and D: Cells were cultured for 48 h with 6 mM NaB or for 24 h with 250 nM UCN-01 or 20 nM STS, alone or in combination followed by Western blotting to detect p53, p21 and p73. Blottings using an anti-β-actin antibody served as protein loading controls. NaB and UCN-01 (C) or STS (D) combination increased the expression of the three cell cycle regulators studied.
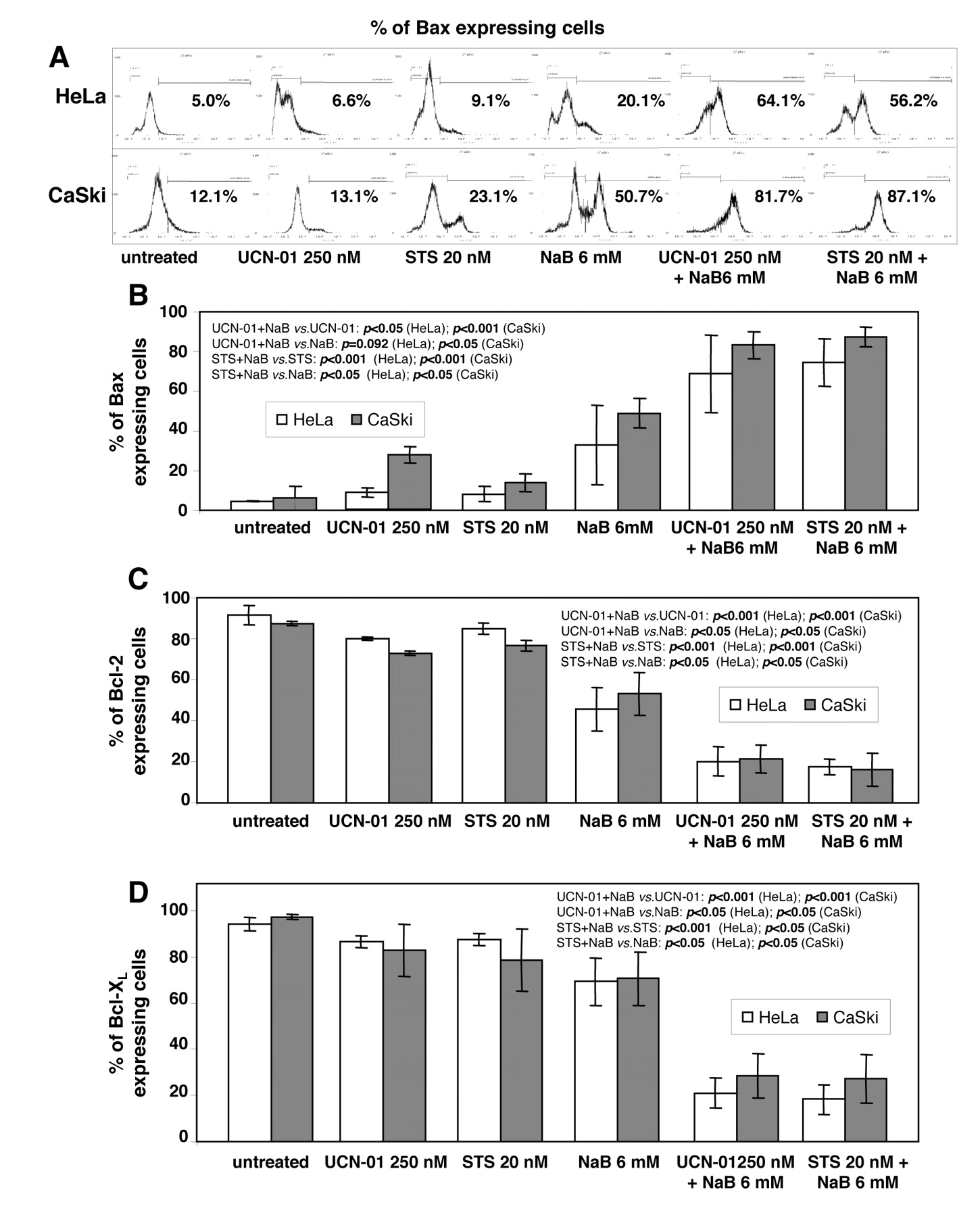
Effect of NaB and UCN-01 or STS on the cellular expression of BAX, BCL-2 and BCL-XL. HeLa and CaSki cells were cultured for 48 h with 6 mM NaB or for 24 h with 250 nM UCN-01 or 20 nM STS, alone or in combination, stained with specific antibodies recognizing BAX (A and B), BCL-2 (C) and BCL-XL (D) and analysed by flow cytometry as described in the Materials and Methods. FACS analyses show repartition of cell mean fluorescence intensity after BAX immunostaining of HeLa and CaSki cells (A) and histograms show the percentage of positive cells for each protein immunostaining (B, C and D). Increased expression of BAX (B) and decreased expression of BCL-2 (C) and BCL-XL (D) were significant in co-treated cells compared to cells treated with a single agent. Data are the means±s.d. of 3 determinations.

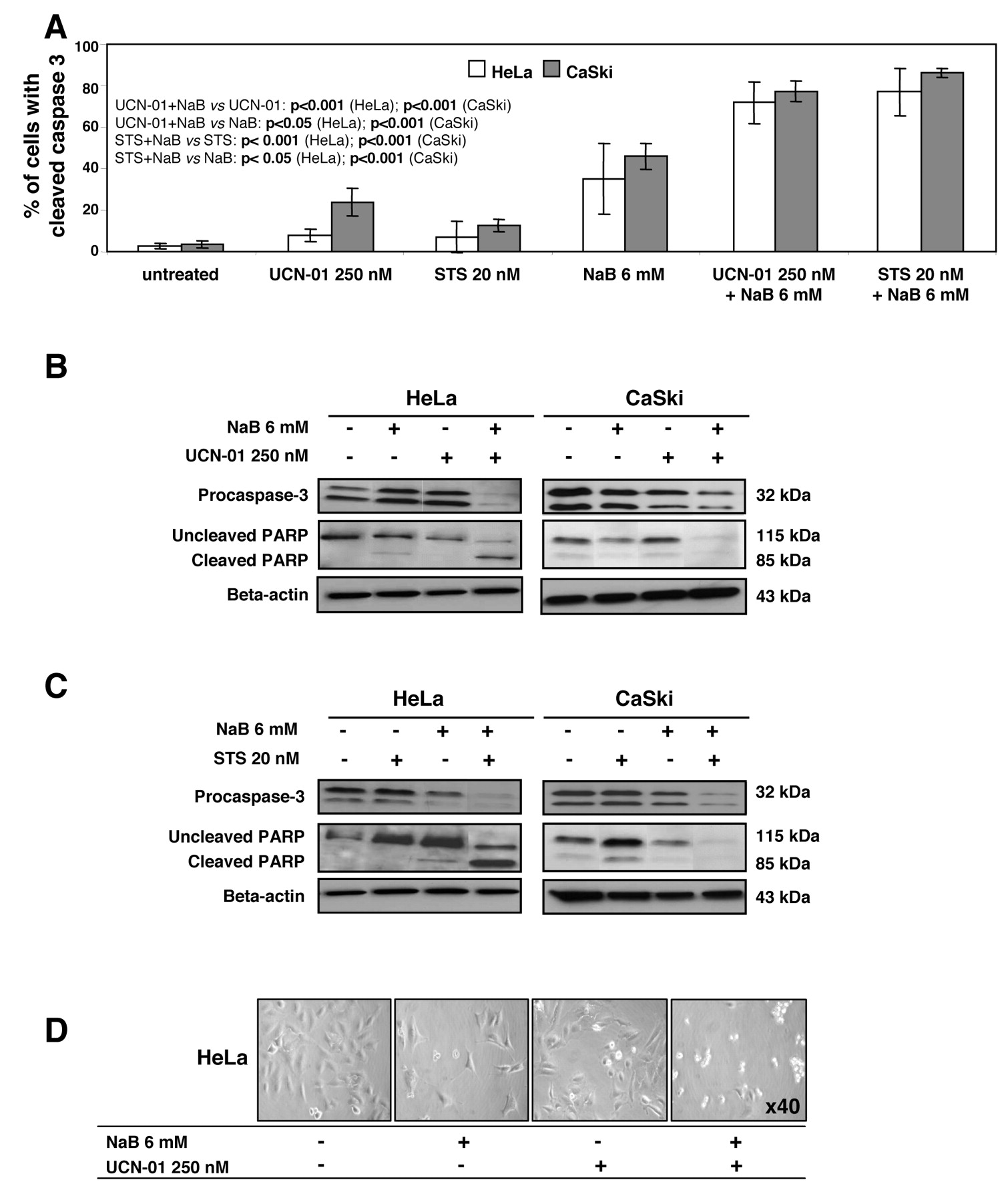
NaB/UCN-01 or STS-mediated cell death is associated with caspase activation and PARP cleavage. HeLa and CaSki cells were cultured for 48 h with 6 mM NaB, or for 24 h with 250 nM UCN-01 or 20 nM STS, alone or in combination. A: Flow cytometric analysis of HeLa and CaSki cells showing cleaved caspase-3 immunostaining after the different treatments. The values indicate the percentage of cells with active caspase-3 which significantly increased in co-treated cells compared to cells treated with a single agent. Data are the means±s.d of 3 determinations. B and C: Western blots of cell lysates were performed and were probed with the corresponding antibodies. They confirm procaspase-3 processing, as well as PARP cleavage, which were more important in cells co-treated with NaB and UCN-01 (B), or STS (C) compared with single treatment. Blottings using an anti-β-actin antibody served as protein loading controls. D: Representative images of HeLa cells after exposure to NaB, UCN-01, or both drugs. More apoptotic cells are visualized after co-treatment compared to single treatment.
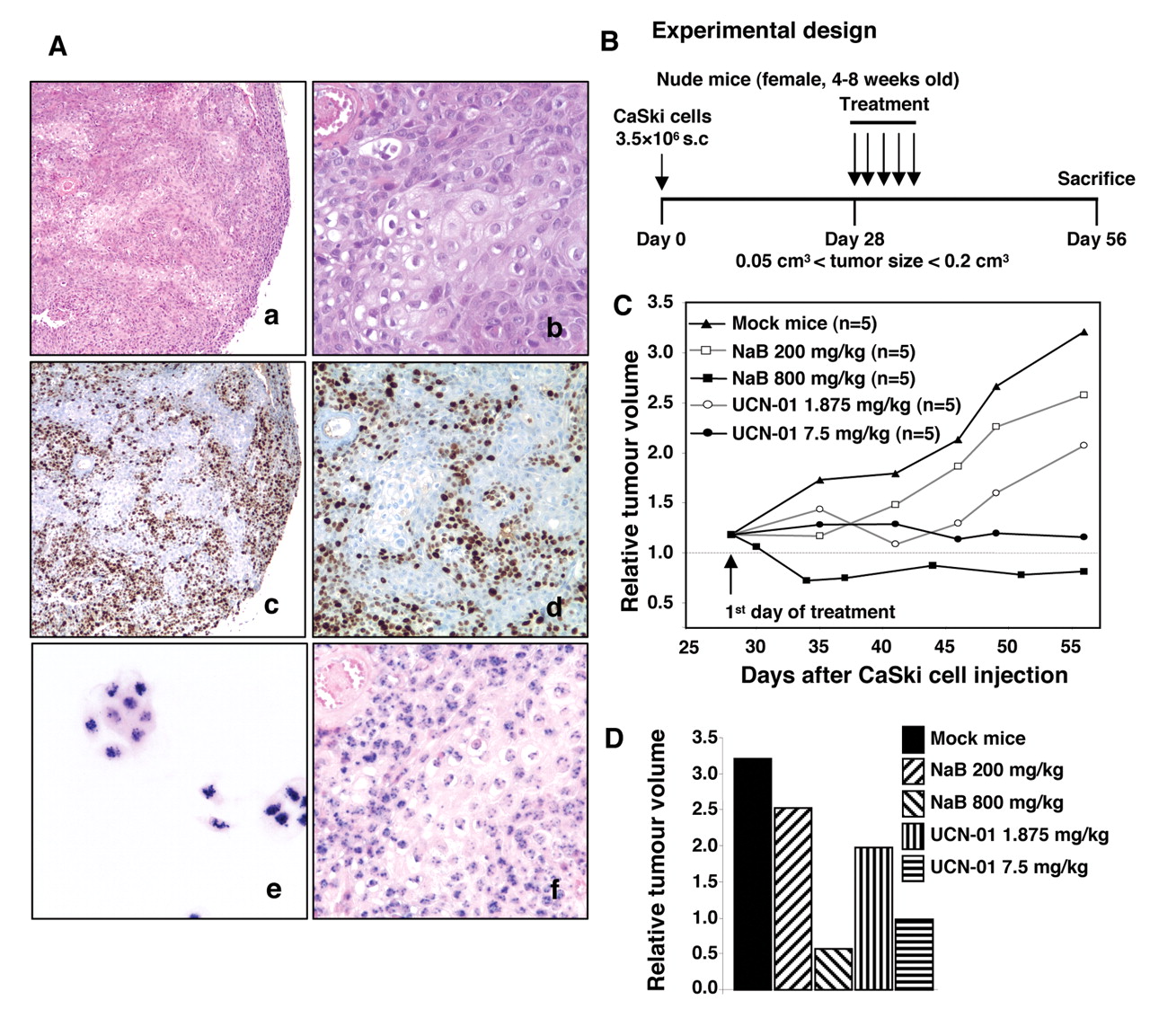
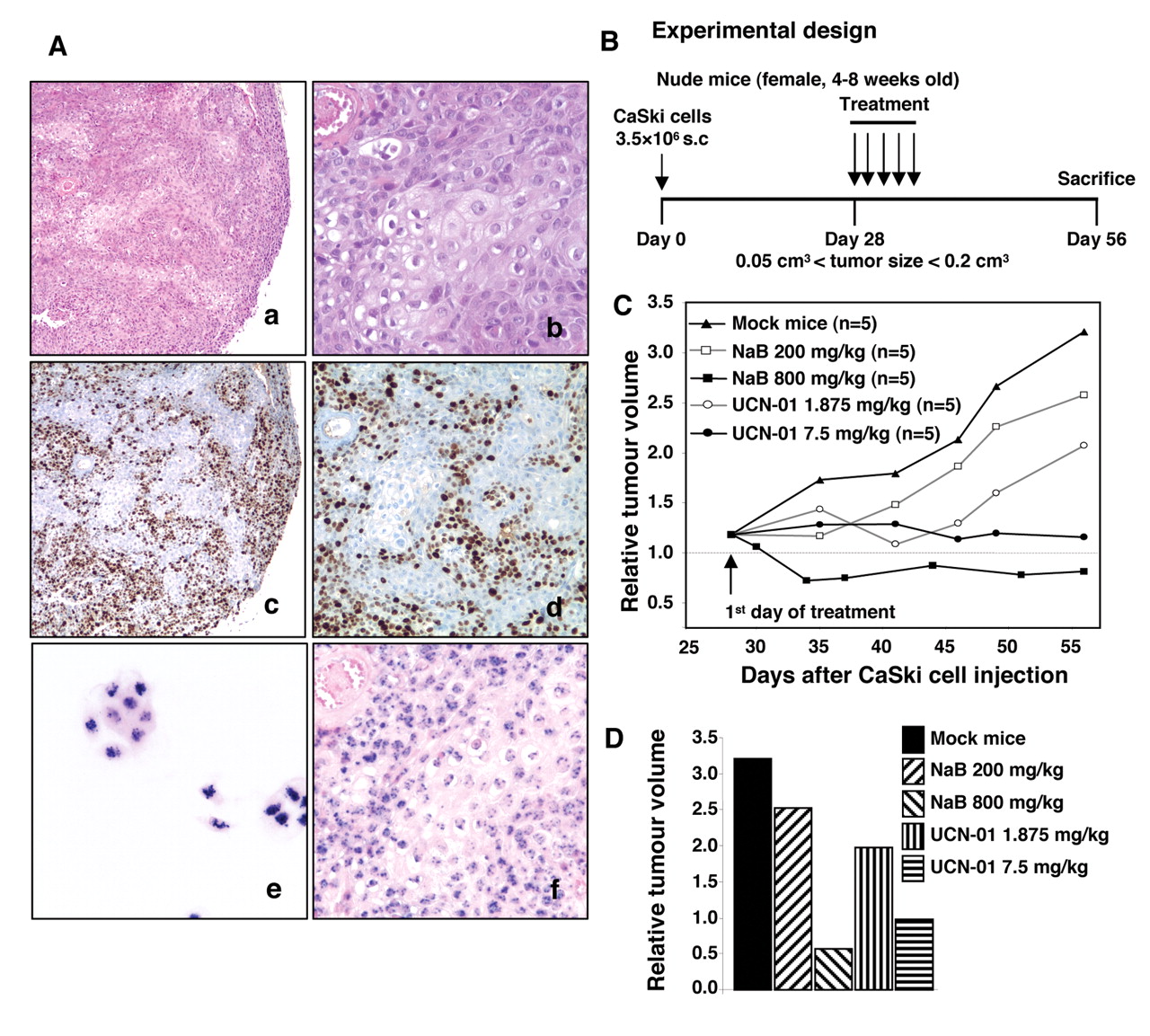
Analysis of xenografted tumours. A: Tumours were dissected, fixed in 4% buffered formalin and paraffin-embedded. Five μm sections were treated for histology (a, b), Ki-67 immunohistochemistry (c, d) and HPV DNA in situ hybridization (e, f). CaSki cells were used as positive control for the latter (e). Microphotographs were taken with ×5 (a, c), ×10 (d) and ×20 objectives (b, e, f). B: Schematic representation of the experimental design: CaSki-bearing mice were treated for 5 days with NaB (200 or 800 mg/kg) or with UCN-01 (1.875 or 7.5 mg/kg) intraperitoneal (i.p.) injection 4 weeks after grafting. Mock mice were treated with vehicle. C: Tumour growth was evaluated by plotting the mean of the relative tumour volume per group (data from each group consisted of 5 mice due to premature death) against time after first treatment. D: Relative tumour volume at the end of the experiment, 4 weeks after treatment beginning.
In view of the above results, studies were performed to determine whether the HDAC/PKC inhibitor regimen resulted in an enhanced induction of p53 and p73 expression. The pretreatment of cells by 6 mM NaB followed by 250 nM UCN-01 or 20 nM STS increased the amounts of these two tumour suppressor proteins (with the exception of p53 in HeLa cells under NaB plus UCN-01), as well as their common target p21 (Figure 2C-D), which might contribute to G1 cell cycle arrest and enhanced apoptosis.
Because cell death is tightly regulated by the balance between pro-apoptotic (e.g. BAX) and anti-apoptotic (e.g. BCL-2 and BCL-XL) BCL-2 family members, we determined what effects combined exposure to NaB and UCN-01 or STS might have on such protein expression. As shown in Figure 3, NaB, UCN-01 and STS given individually at low doses had moderate effects on the expression of BAX, BCL-2 and BCL-XL, while when used in combination, they led to significant increases in BAX levels and pronounced decreases in BCL-2 and BCL-XL.

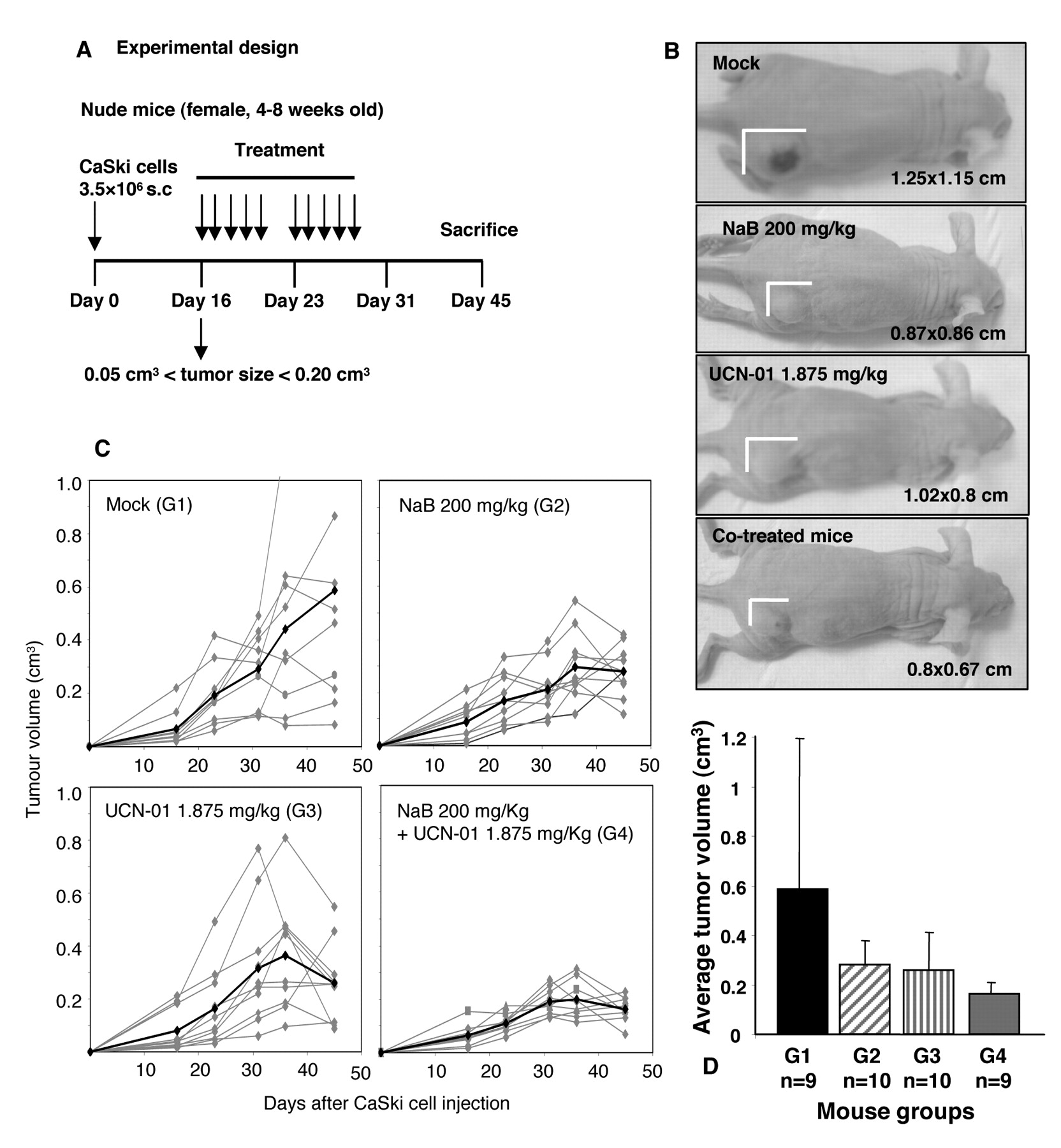
Tumour volumes in NaB-and UCN-01-treated nude mice bearing subcutaneous CaSki cell tumours. A: Schematic representation of the experimental design: mice bearing 0.05-0.20 cm3 s.c. tumours were treated with vehicle (G1, mock mice), NaB at 200 mg/kg (G2, NaB), UCN-01 at 1.875 mg/kg (G3, UCN-01), or with both using NaB at 200 mg/kg plus UCN-01 at 1.875 mg/kg (G4, combination) delivered i.p. 5 times a week for 2 consecutive weeks. B: Outward appearance of CaSki cell tumours in nude mice with different treatments. C: Tumour growth curves of CaSki xenografts as a function of time. The grey lines represent the change in tumour volume for each mouse. The black line represents the average tumour volume of the group. D: Average tumour volumes at the end of the experiment, 4 weeks after beginning of treatment.
Cell death mediated by combination of NaB with UCN-01 or with STS involves activation of caspase-3 leading to PARP cleavage. Immunostaining of cleaved caspase-3 followed by flow cytometric analysis, as well as Western blot analysis of lysates obtained from cells exposed to NaB, UCN-01, or STS alone showed minimal cleavage of procaspase-3 and modest degradation of the caspase-3 substrate PARP. However, sequential exposure of cells to NaB followed by UCN-01 or STS revealed a marked induction of caspase-3 activation and degradation of PARP (Figure 4A-C). Thus NaB or UCN-01 alone was minimally toxic, but the combination yielded a marked increase in cell death, as shown by rounding of the cells with plasma membrane blebbing (Figure 4D).
CaSki tumour cell growth in nude mice. CaSki xenograft-derived tumours consisted of a solid mass containing nests of undifferentiated cells with hyperchromatic nuclei, high nuclear/cytoplasmic ratio and abnormal mitotic figures. Moreover, vascularization was present, which may contribute to tumour development (Figure 5A, a-b). The in vivo proliferation of xenografted CaSki cells was confirmed by a strong nuclear Ki-67 immunostaining (Figure 5A, c-d). The in situ hybridization allowed the detection of HPV16 DNA in cultured CaSki cells (Figure 5A, e) and xenograft tissue sections (Figure 5A, f); the staining appeared either as many dots or as a combination of diffuse and punctuated dots, as we previously described (38).
Due to its clinical toxicity, STS was not used in animal models. Furthermore, experiments were performed to define an optimal schedule for drug administration enabling NaB and UCN-01 to be administered at lower dosages (Figure 5B). Treatment with 800 mg/kg/day NaB or 7.5 mg/kg/day UCN-01 for 5 consecutive days did not affect the body weight of the mice (data not shown), while the tumour volumes at the end of the experiments were significantly lower than in the mock-treated mice (Figure 5C-D). When mice were treated with 200 mg/kg/day NaB or 1.875 mg/kg/day UCN-01, the tumour volumes were reduced by 35% and 50% respectively compared with control animals (Figure 5C-D).
After these pilot studies were carried out, we next determined whether UCN-01 could potentiate the antitumour effects of NaB in nude mice (Figure 6A). As illustrated in Figures 6B-D, the volume of the tumours of mice treated with the two compounds (G4) was significantly reduced as compared with monotherapy groups (G2, G3) or mock-treated mice (G1). Our data provide further evidence indicating that such a therapeutic regimen may have significance in application for cervical cancer chemotherapeutic purposes.
Discussion
Ten years ago, several randomized trials including almost 1800 patients with locally advanced cancer of the cervix demonstrated a survival benefit of 30-50% for cisplatin-based chemoradiation compared with radiotherapy alone. However oncologists are still concerned about the efficacy and toxicity of cisplatin-based chemoradiation (39). Our interest in the combination of NaB and UCN-01, both agents already introduced into clinical trials, stemmed from reports showing that HDACi could increase tumour cell susceptibility to chemotherapeutic agents or ionizing radiations (7). Moreover, combination of different antitumour treatment modalities provides insights into their optimum use to achieve clinical benefits by limiting unspecific toxicity often observed for the use of an exceedingly high dose single treatment regimen.
Recently, Finzer et al. found that NaB activates an intrinsic death program in HeLa cells by inducing pro-apoptotic forms of p73 (17), while previous results in our group showed that STS triggers intrinsic apoptosis of HeLa and CaSki cells through a p53-dependent pathway (27). The present findings indicate that in these HPV-positive cells derived from cervical carcinomas, UCN-01 potentiates antiproliferative and cytotoxic activities of NaB at much lower concentrations than that required to activate apoptosis by each individual drug alone. Indeed, the combined administration of NaB with UCN-01 potently enhanced mitochondrial dysfunction and caspase-3 activation leading to PARP degradation and apoptosis. These events are associated with changes in cell cycle and apoptotic regulatory proteins. This is in agreement with a previous study showing that a low concentration of butyrate that itself did not induce activation of caspase-3 or apoptosis rendered Jurkat lymphoid and LIM1215 colorectal cancer cells highly susceptible to induction of apoptosis by STS (22), an agent that acts by causing mitochondrial release of cytochrome c (30). In addition in those cells, activation of caspase-3 induced by butyrate-primed cells enhanced caspase-3 activation by more than 7-fold by addition of cytochrome c and dATP to isolated cytosol (22).
The mechanism by which NaB renders HeLa and CaSki cells more susceptible to apoptotic stimuli induced by UCN-01 exposure is not clear. The main problem in elucidating the mechanisms of action of HDACi and protein kinase inhibitor is their pleiotropic effects that involve multiple signalling pathways. Although the effect of HDACi on histone proteins is well understood, many non-histone proteins have been identified as substrates for HDAC (40). As depicted in Figure 2, in the combined regimen, there was an accumulation of the two pro-apoptotic proteins, the transcriptionally active TAp73 and p53. The p73 accumulation results very likely from the acetylation of the transcription factor E2F1 that promotes its recruitment on the p73 promoter and its transcriptional activation as previously shown by Finzer et al. in HPV-positive cells treated with NaB (17). Moreover, the E6 oncoprotein of HPV that is inhibited in HeLa and CaSki cells under STS exposure as shown in our previous study (29), was unable to abrogate the co-stimulatory function of p300/CBP, which could also favor p73 accumulation (41). Similarly to p73, we may speculate that NaB induced acetylation of p53 and the acetylated p53 acquired stability and increased transactivation activity (42). Moreover, MDM2 would be unable to promote p53 degradation, since HDAC1 necessary for p53 deacetylation is also likely inhibited (43). It is also conceivable that the p53 level is increased due to the reduction of MDM2 and E6 oncoprotein under protein kinase inhibitor exposure as we previously observed (29).
Although nearly half of the genes transcriptionally induced by p53 and p73 overlap (44), here the overexpression of common targets p21 and BAX genes can partly explain G1 growth arrest and apoptosis of cervical cancer cells in presence of NaB plus UCN-01. Moreover, down-regulation of pro-survival BCL-2 and BCL-XL proteins (Figure 3) participates in the commitment of cells to apoptosis. Whether these members of the BCL-2 family can control the mitochondrial death pathway, p53 likely plays an apoptogenic role at mitochondria through direct activation of BAX (45) or through binding to BCL-2 and BCL-XL which blocks their activity (45, 46), leading to cytochrome c release and caspase activation. The ability of p73 to induce apoptosis could be mediated through the direct transcription of BAX itself, as well as through the transcriptional activation of p53 up-regulated modulator of apoptosis (PUMA), which was originally characterized as a p53 target and which facilitates mitochondrial translocation of BAX (47).
The aim of this work was also to establish xenografts of human cervix-derived carcinoma cells transplantable to immunocompromised nude mice. Such a preclinical model could help to test the efficacy of new antitumour drugs and new combinations. Our experiments show that the subcutaneously implantation provides high tumour take, however, with interindividual differences. Importantly, the in situ architecture was inherited by the xenografts and cell proliferation was confirmed by the Ki-67 immunostaining. Moreover, comparison of the HPV status of the original CaSki cells and their xenografts was established by in situ hybridization. HPV DNA positivity was found to be similar, confirming that the xenografts are purely tumoural.
Responses of xenografts to NaB and UCN-01 have been tested in several experimental conditions. Preliminary experiments showed that our strain of nude mice were able to tolerate up to 800 mg/kg of NaB and 7.5 mg/kg UCN-01 for 5 days (Figure 5C-D) with no apparent side-effects such as body weight loss. Interestingly, the combination of NaB and UCN-01 at lower concentrations had better antitumor efficacy than either agent alone, which is consistent with our in vitro findings. The tumour growth inhibition might be due to increased cell cycle arrest and apoptosis but also to the anti-angiogenic properties of HDACi (13, 48).
In this report, we have presented for the first time complementary in vitro and in vivo data on the effects of NaB combined with UCN-01 on cervical carcinoma cell lines HeLa and CaSki. Both drugs alone at low doses had minimal antitumoral activity, but when used in combination, they significantly induced apoptotic cell death and enhanced tumour growth control. Altogether, these results support the potential benefit of the association of NaB and UCN-01 for promoting death of cervical carcinoma cells through p53 and p73 signalling intrinsic apoptotic pathways. Darvas et al. very recently provided new evidence that HeLa cells can be highly sensitized to tumour necrosis factor (TNF) alpha and TNF-related apoptosis inducing ligand (TRAIL) after HDAC inhibition and E7 viral oncogene may represent the link between intrinsic and extrinsic apoptosis (49).
Acknowledgements
The Authors are grateful to Institut National du Cancer (INCa) France, German Academic Exchange Service (DAAD), Germany, Région de Franche-Comté and Ligue Contre le Cancer (Département du Doubs) France for financial support. AZDB was a recipient of a post-doctoral fellowship from INCa, MLP and MN were recipients of pre-doctoral fellowships from INCa and the city of Besançon respectively.
- Received July 13, 2010.
- Revision received August 23, 2010.
- Accepted September 7, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.