


Abstract
Background: 1α,25-dihydroxyvitamin D3 [1,25-(OH)2D3] is the biological active form of vitamin D. Its antiproliferative capacities make it a potential drug to treat diseases such as cancer. The clinical use of 1,25-(OH)2D3 as an antiproliferative agent is hampered by its calcemic effects. Hence, structural analogs such as the seco-9,11-bisnor-17-methyl analog, WY1112, have been developed with superagonistic capacities. This study aims to distinct the molecular activities of 1,25-(OH)2D3 and WY1112 and identify possible differences in gene expression. Materials and Methods: Total RNA was extracted from MCF-7 breast cancer cells treated with 1,25-(OH)2D3 or WY1112 and was used for microarray analysis. Results: The experiments revealed that WY1112 induces the same genes as 1,25-(OH)2D3, but the induction level of the individual genes is higher. Microarray analysis did not reveal genes that were exclusively regulated by WY1112. Conclusion: The superagonistic vitamin D analog WY1112 induces the same set of genes as 1,25-(OH)2D3, but the level of induction of the individual genes is higher.
The human body is capable of producing the major portion of vitamin D3 itself by photosynthesis in the skin. Under the influence of UV-B light, it converts 7-dehydrocholesterol to previtamin D3. Thermal energy causes this unstable metabolite to isomerize to vitamin D3 (1). Moreover, fatty fish and fortified dairy products are important dietary sources of vitamin D3. Two sequential hydroxylation steps convert vitamin D3 to its biologically most active form, 1α,25-dihydroxyvitamin D3 [1,25-(OH)2D3] (2).
1,25-(OH)2D3 exerts its effects via the vitamin D receptor (VDR), which belongs to the steroid/thyroid hormone receptor family (3). After dimerization of the VDR with the retinoid X receptor (RXR) receptor, the complex binds to vitamin D response elements (VDREs), recruits co-regulators and the transcriptional preinitiation complex to initiate target gene transcription (4-5). 1,25-(OH)2D3 plays a crucial role in bone metabolism and in mineral homeostasis (classical effects) but has also potent antiproliferative and prodifferentiating actions on normal as well as malignant cell types (non-classical effects) [reviewed in (6)]. This potent growth inhibitory effect, combined with the presence of the VDR in a wide variety of cells, makes 1,25-(OH)2D3 an ideal compound to treat hyperproliferative disorders such as cancer.
Calcemic effects, however, hamper therapeutic application of 1,25-(OH)2D3 in hyperproliferative diseases. Consequently, a lot of effort has been put into the design of structural analogs of 1,25-(OH)2D3 with increased antiproliferative capacity and reduced calcemic effects [reviewed in (7)].
Some years ago, we embarked an extensive study of the structure-function relationship with focus on the central CD region of 1,25-(OH)2D3. 1,25-(OH)2D3 was stripped down to its five-carbon backbone (C8-C20) and resubstituted in various ways. WY1112 is such a non-steroidal analog of 1,25-(OH)2D3, lacking the full six-membered C-ring (D-ring analog). Analogs that have similar alterations have improved antiproliferative potency (8). This seco-9,11-bisnor-17-methyl analog of 1,25-(OH)2D3 possess a 20-epi side chain that is fluorinated at C26 and C27 (Figure 1), modifications which have been shown to make analogs more resistant to degradation and improve their halflife, usually resulting in analogs with more potent antiproliferative actions, but also stronger calcemic effects (9). The epimerization of C20 is also an important modification which reorients the six-carbon side chain of 1,25-(OH)2D3 substantially and alters the metabolic degradation (10). This analog is 400-fold more potent than 1,25-(OH)2D3 in reducing cell growth, but has similar calcemic effects.

Chemical structure of 1,25-(OH)2D3 and analog WY1112.
The molecular mechanism of how 1,25-(OH)2D3 or the analogs exert their antiproliferative action is not completely understood. The present study aims to unravel the molecular mechanisms behind the superagonistic vitamin D-analogs. WY1112 was selected because of its very potent antiproliferative action. Its molecular mode of action was studied through comparison of gene expression profiles obtained by extensive microarray studies performed on MCF-7 breast cancer cells at different time points after treatment with a single dose of 1,25-(OH)2D3 or WY1112.
Materials and Methods
Cell culture. MCF-7 cells (American Type Culture Collection (ATCC), Rocheville, Maryland, USA) were maintained in DMEM with 2 mM glutaMAX™-I (Invitrogen, Merelbeke, Belgium) containing 10% fetal bovine serum (Biochrom KG, Berlin, Germany), 100 units/ml penicillin and 100 μg/ml streptomycin.
Proliferation assay. To measure cellular proliferation, MCF-7 cells (1×104 cells/well) were seeded in 96-well dishes. After a 72-h incubation period with increasing concentrations of 1,25(OH)2D3 or WY1112, 1 μCi [3H]thymidine (specific activity of 2 Ci/mmol) was added. Cells were harvested after an additional 4- to 6-h incubation period.
Cell cycle analysis. At 24, 36 and 48 h after treatment with 3×10-8 M 1,25-(OH)2D3 or vehicle (ethanol) alone, approximately 1×106 MCF-7 cells were washed with PBS twice and fixed in ice-cold 70% ethanol for 30 min. After fixation, cells were washed twice with PBS containing 0.05% Tween-20 and resuspended in PBS containing 0.05% Tween-20, 0.5 mg/ml propidium iodide and 1 mg/ml RNase A (Sigma-Aldrich, NV/SA, Bornem, Belgium). Analysis of samples was done using the CellQuest and Modfit program on a FACSort flow cytometer (Becton Dickinson, Erembodegem, Belgium).
Binding affinity. The affinity of WY1112 to the VDR was evaluated by its ability to compete with [3H]1,25-(OH)2D3 for binding to high-speed supernatant from intestinal mucosa homogenates obtained from normal pigs. The incubation was performed at 4°C for 20 h, and phase separation was obtained by the addition of dextran-coated charcoal. The relative affinity of the analogs was calculated from their concentration needed to displace 50% of [3H]1,25-(OH)2D3 from its receptor compared with the activity of 1,25-(OH)2D3 (assigned a value of 100%).
Binding of WY1112 to human serum vitamin D-binding protein (hDBP) was performed at 4°C. [3H]1,25-(OH)2D3 and 1,25-(OH)2D3 or WY1112 were added in 5 μl of ethanol into glass tubes and incubated with hDBP (0.18 μM) in a final volume of 1 ml (0.01 M Tris-HCl buffer and 0.154 M NaCl, pH 7.4) for 3 h at 4°C. Phase separation was then obtained by the addition of 0.5 ml of cold dextran-coated charcoal.
RNA isolation. For the microarray analysis, the MCF-7 cells were treated with a concentration of 3×10-8M 1,25-(OH)2D3, WY1112, or vehicle alone.
Cells were harvested at 1 h, 3 h, 6 h, 12 h, 24 h and 36 h after the treatment in two independent experiments. Total RNA from MCF-7 cells used for microarray analysis was prepared using the RNeasy kit (Qiagen, Hilden, Germany). The RNA extraction method was performed as specified by the manufacturer.
Microarray analysis. Microarray analyses were performed at the Microarray Facility of the Flanders Interuniversity Institute for Biotechnology (VIB). A Human Genome U133 Plus 2.0 array (Affymetrix, Inc., Santa Clara, CA, USA) was utilized for each sample.
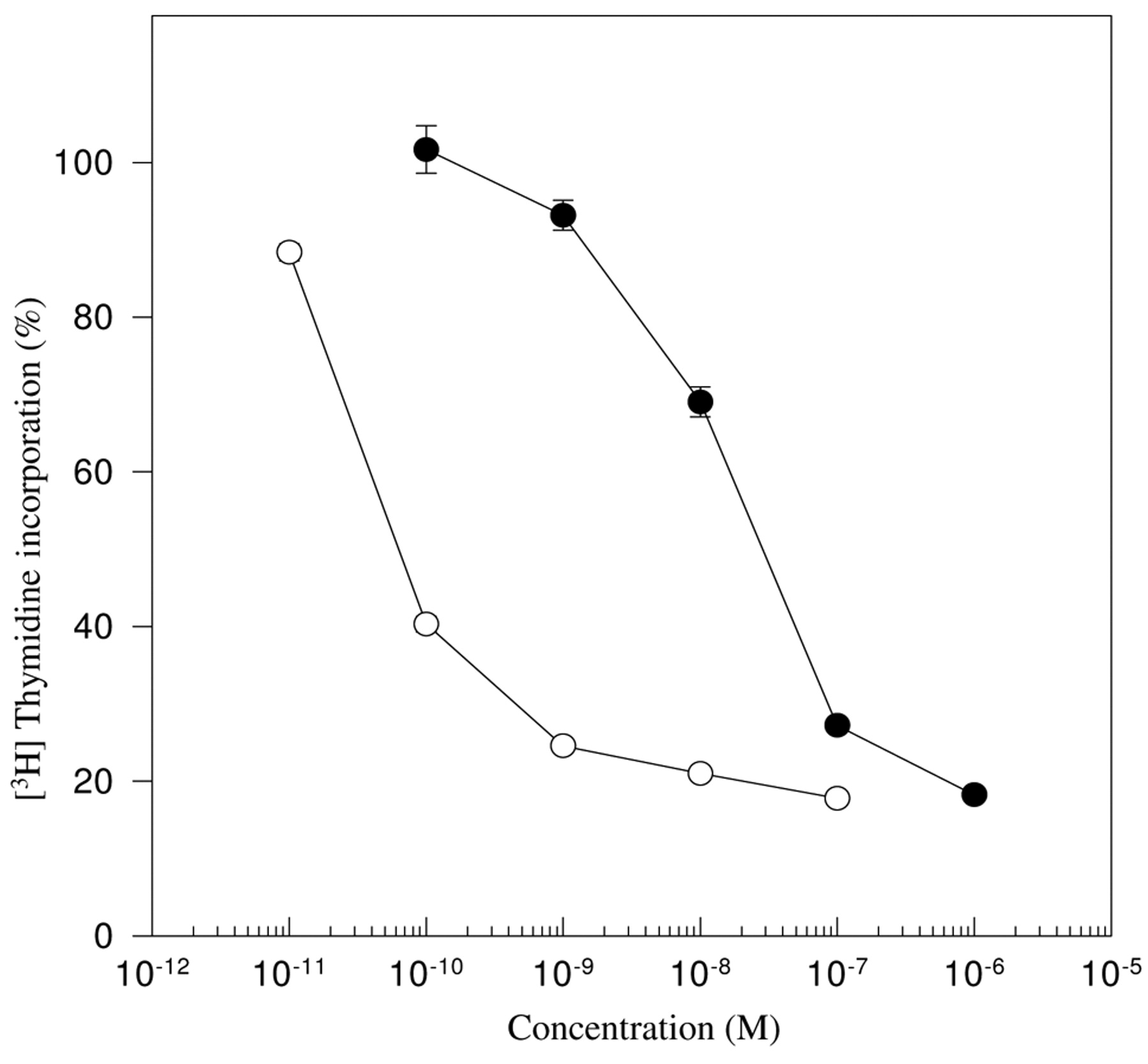
Effects on MCF-7 cell growth. A [3H]thymidine incorporation assay was performed on MCF-7 cells. Cells were treated with different concentrations of 1,25-(OH)2D3 (●) and WY1112 (○) for 72 h prior to the measurement of [3H]thymidine incorporation.
Results
Antiproliferative capacity of WY1112. The EC50 value, the concentration at which [3H]thymidine incorporation is reduced to 50%, was 3×10-8M for 1,25-(OH)2D3 and 7×10-11M for WY1112 (Figure 2).
1,25-(OH)2D3-induced growth inhibition is characterized by a blocked transition from G1 to S-phase of the cell cycle, causing the cells to accumulate in the G1-phase. MCF-7 cells were treated with 3×10-8M 1,25-(OH)2D3, WY1112, or vehicle (ethanol) alone. This dose reduced growth of 1,25-(OH)2D3-treated and WY1112-treated MCF-7 cells by 50% and 80% respectively according to the [3H]thymidine incorporation assay (Figure 2).
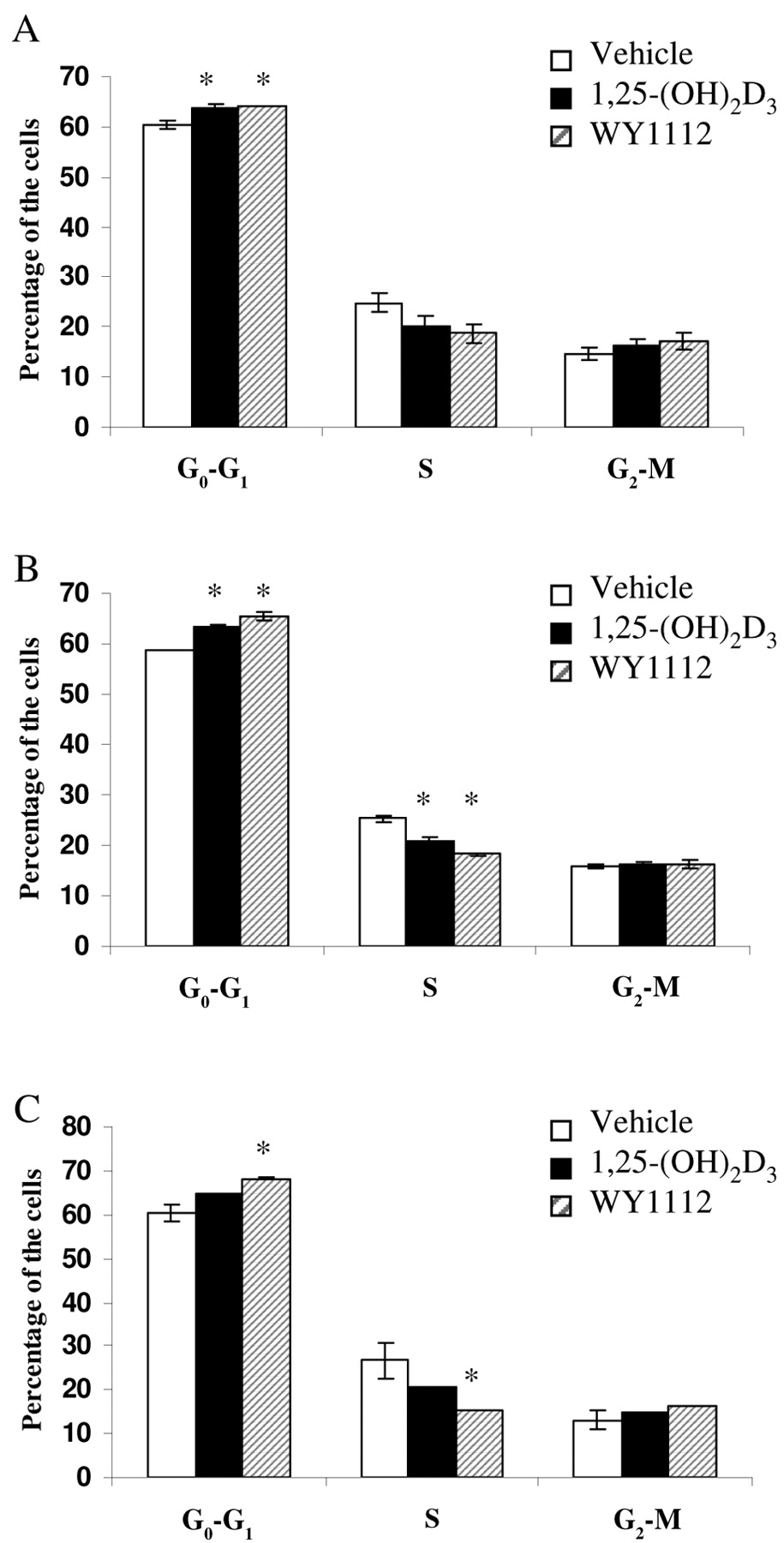
At 24 h after treatment, there was no significant drop in the number of S-phase cells. In contrast, at 36 h, samples contained 5% (1,25-(OH)2D3) or 7% (WY1112) fewer S-phase cells. A treatment of 48 h reduced the proportion of cells in the S-phase by 6% (1,25-(OH)2D3) and 11% (WY1112) (Figure 3).
Binding affinity. The affinity of 1,25-(OH)2D3 for hDBP was 2.2×107 M-1, whereas its affinity for the pig duodenal mucosa VDR was 1.3×1010 M-1. The affinity of WY1112 for hDBP was 6.8×105 M-1, which is only 2% compared to 1,25-(OH)2D3. Moreover, the affinity of WY1112 for the VDR was 1.1×109 M-1, still 20% of the 1,25-(OH)2D3-affinity (Figure 4).

Effects of 1,25-(OH)2D3 and WY1112 on the cell cycle. FACS analysis of MCF-7 cells at 24 h (A), 36 h (B) and 48 h (C) after treatment with 3×10-8M 1,25-(OH)2D3 or WY1112 or vehicle (ethanol) alone. *Significantly different from control treated cells at 0.05 significance level according to Scheffe's multiple comparison test.
Microarray study. A cluster analysis, which was performed to group genes that have similar expression patterns, revealed the presence of 19 significantly different clusters. One of these clusters contained 142 genes, which were up-regulated both by 1,25-(OH)2D3 and by WY1112. All the other clusters contained genes of which the temporal expression profile did not differ significantly between vehicle-, 1,25-(OH)2D3- and WY1112-treated cells. Although WY1112 is a superagonistic analog, with a 400-fold stronger antiproliferative capacity than 1,25-(OH)2D3, this compound does not regulate a different set of genes. The same genes are induced by both compounds, although the induction capacity of WY1112 is higher than that of 1,25-(OH)2D3.
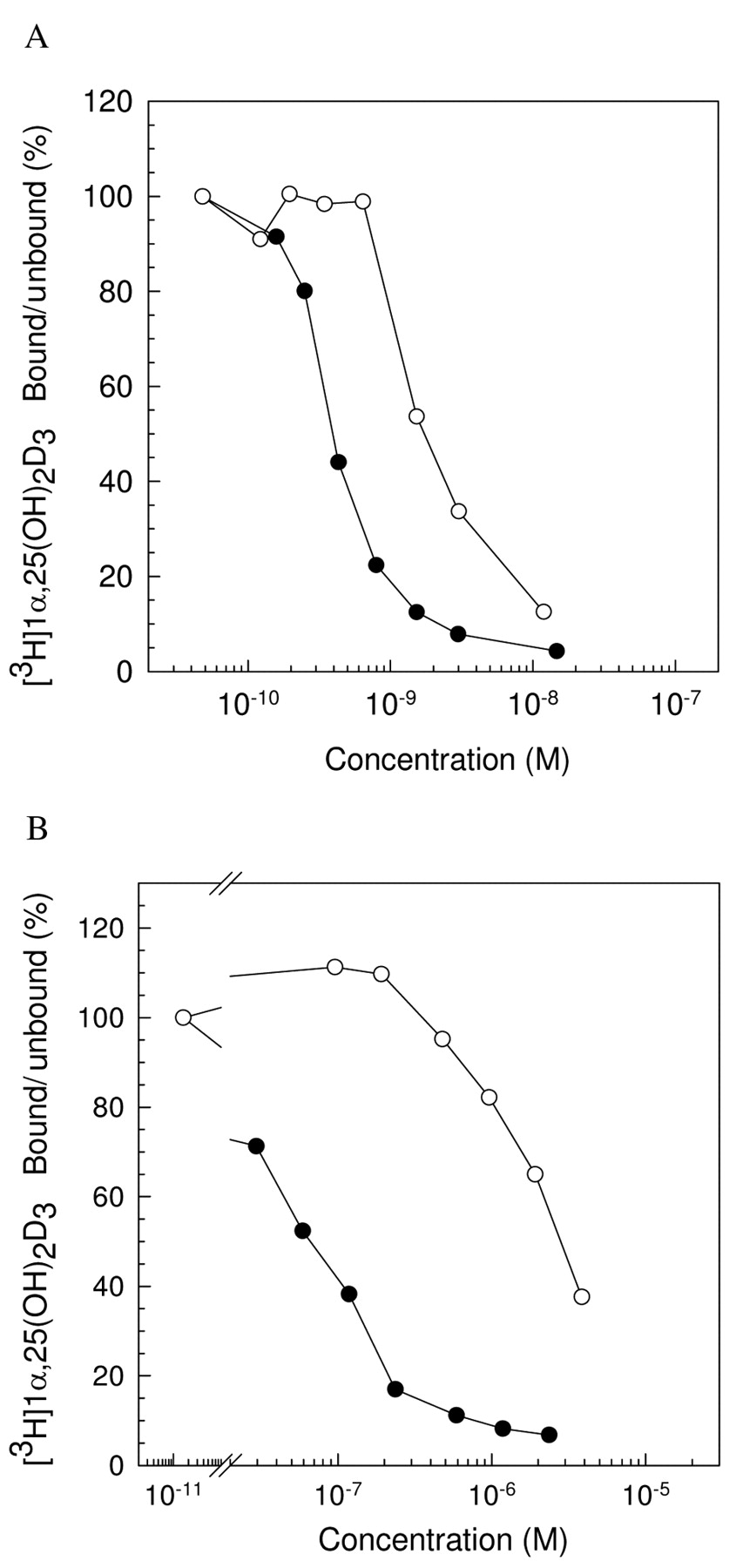
WY1112 binding affinity. MCF-7 cells were treated with different concentrations of 1,25-(OH)2D3 (●) and WY1112 (○) in the presence of [3H]1,25-(OH)2D3. Binding of [3H]1,25-(OH)2D3 to the VDR (A) or DBP (B) was measured.

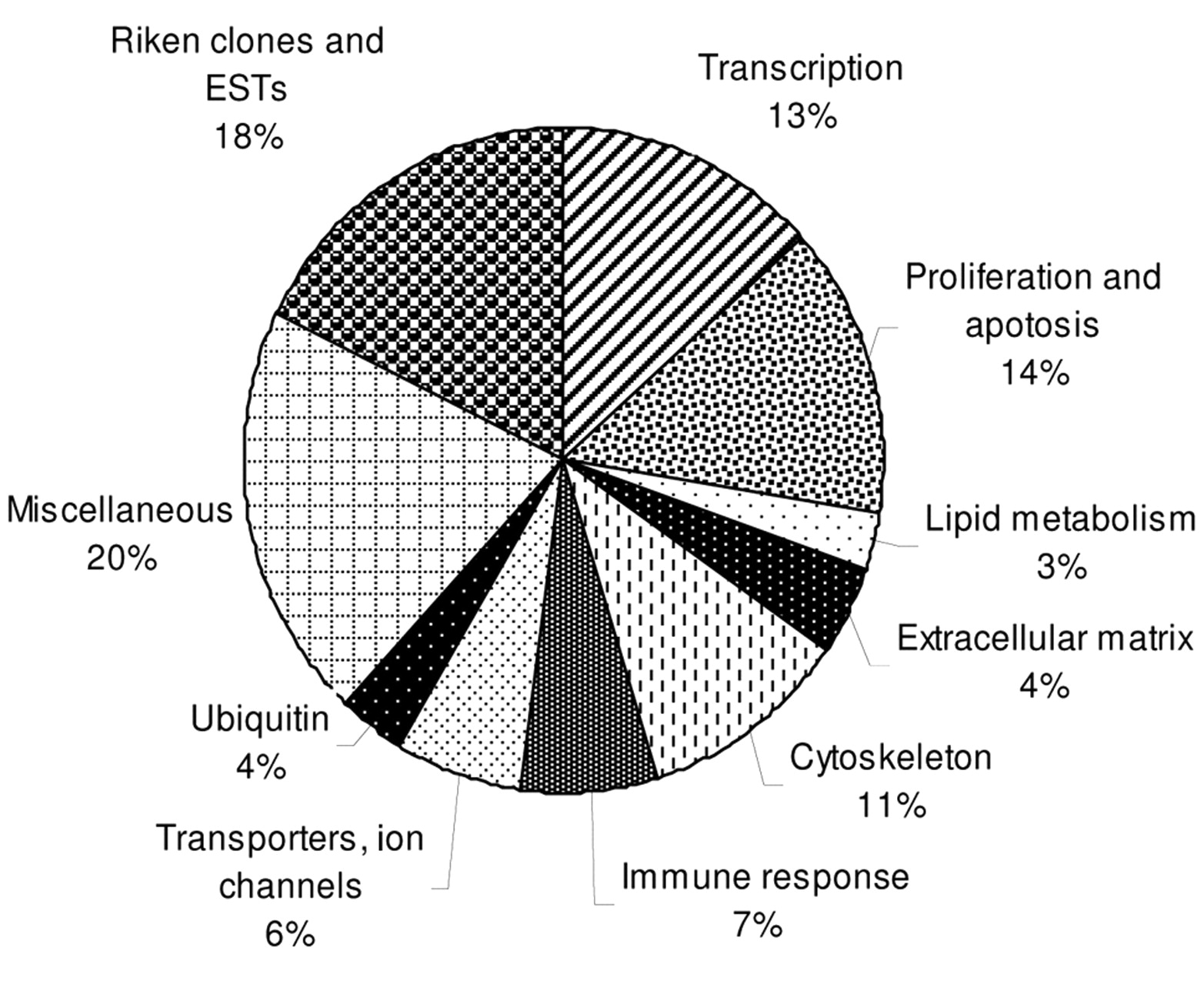
Clusters of up-regulated genes.
Genes that were up-regulated can be divided into groups that are involved in proliferation and apoptosis (14%), gene transcription (13%), cytoskeleton organization (11%), immune response (7%), transmembrane transport (6%), ubiquitination (4%), extracellular matrix (4%) and lipid metabolism (3%) (Figure 5).
Discussion
The list of genes that were up- and down-regulated following 1,25-(OH)2D3 treatment includes a number of known 1,25-(OH)2D3 target genes. The most responsive gene identified was 24-hydroxylase. This mitochondrial enzyme is responsible for inactivating vitamin D metabolites through the C-24 oxidation pathway. Genes and pathways reported to be regulated by 1,25-(OH)2D3 in MCF-7 cells were also affected in our microarray study. Apoptosis and cell-cycle-arrest genes, such as Bcl2-associated X protein (Bax) and growth arrest- and DNA damage-inducible gene 45 alpha (GADD45α) for instance, are known targets of 1,25-(OH)2D3 in MCF-7 cells that were also affected in this study (11). A number of growth factor genes were also regulated, namely insulin-like growth factor-binding protein 3 (IGFBP3), transforming growth factor beta-2 (TGFβ2), epidermal growth factor receptor (EGFR) and dual-specificity phosphatase 10 (DUSP10) (11, 12). Moreover, the treatment of 1,25-(OH)2D3 also induced the expression of some oncogenes such as jun B proto-oncogene, v-fos FBJ murine osteosarcoma viral oncogene homolog (Fos) and GTP-binding protein overexpressed in skeletal muscle (GEM) as previously reported (11, 12).
The negative modulation of gene transcription was restricted to 12 genes in this study. Among these genes are components involved in matrix degradation, such as A desintegrin-like and metalloproteinase with thrombospondin type 1 motif 9 (ADAMTS9) and desintegrin-like. The number of genes down-regulated by 1,25-(OH)2D3 is commonly lower than the number of up-regulated genes. Towsend et al. for instance, reported 91 up-regulated versus 5 down-regulated genes in a microarray study performed on MCF-7 cells, treated with 1,25-(OH)2D3 (11). Another microarray study on 1,25-(OH)2D3-treated-MCF-7 cells published 51 up-regulated genes and 19 down-regulated genes (12).
In this microarray study, we also confirmed the regulation of genes that were affected by 1,25-(OH)2D3 in other breast cancer cell lines. In SC3 mammary carcinoma cells for example, GPRK5, a G-protein coupled receptor kinase, was reported to be a 1,25-(OH)2D3 target gene (13). 1,25-(OH)2D3 target genes in MCF10 cells were also confirmed in our microarray. These genes include CD14 antigen, CD97 antigen, serine protease kalikrein 6 (KLK6), metabotropic glutamate receptors Homer homolog 3, calmin and inositol triphosphate receptor 1 (IPTR1) (14).
Despite its highly increased antiproliferative capacity, and its ability to block a higher percentage of the cells in the G1 phase of the cell cycle, WY1112 has the same expression profile as 1,25-(OH)2D3. The fold induction of individual genes is however higher. This finding is in agreement with an earlier published study in which it was reported that the analog EB1089 does not result in gene-specific transactivation compared to 1,25-(OH)2D3 in squamous carcinoma cells (SCC25) (15). EB1089 is a well-characterized vitamin D analog which is 10-fold more potent than 1,25-(OH)2D3 in regulating growth in SCC25 cells (16). The data suggested that differences in action of EB1089 and 1,25-(OH)2D3 arise more from their relative sensitivities to metabolism than from differing effects on VDR function (15). Another microarray study also reported on the gene expression changes induced by the vitamin D gemini analog RO-3582 [1α,25-dihydroxy-20S-21(3-hydroxy-3-methyl-butyl)-23-yne-26,27-hexafluorocholecalciferol] on early premalignant MCF10AT1 and fully malignant MCF10AT1 cells (14). According to their study, RO-3582 regulates many genes differently in the two cell lines, with more significant gene changes in the early premalignant cell line.
In conclusion, WY1112 is a 400-fold more potent antiproliferative agent than 1,25-(OH)2D3. However, both compounds affect the same set of genes. The difference in antiproliferative capacity is consequently not due to a difference in gene regulation. Previous studies already demonstrated a strong correlation between the antiproliferative potency of an analog and its ability to induce VDR-coactivator interactions (17, 18). Altered coactivator binding by the VDR may explain the superagonistic capacity of WY1112 as well. Moreover, it was shown for 14-epi-analogs TX522 and TX527 that modification in the analog's sensitivity to metabolism partly accounts for its improved antiproliferative potential (17). Therefore, this mechanism could be another basic principle of the superagonistic action of WY1112.
Acknowledgements
This work was supported by grants from the Fund for Scientific Research (FWO-G.0587.09 and FWO-G.0553.06) and the KU Leuven Research Council (EF/05/007 SymBioSys). GE is a postdoctoral researcher for the Fonds voor Wetenschappelijk Onderzoek (FWO).
- Received January 29, 2009.
- Revision received May 11, 2009.
- Accepted June 8, 2009.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}