


Abstract
The active vitamin D3, 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3), is now well recognized as a potent regulator of cell proliferation and differentiation in addition to possessing a regulatory effect on calcium and phosphorus metabolism. From research on the synthesis of 1,25(OH)2D3 analogs with the goal of separating these biological activities, we have already reported two characteristic analogs of active vitamin D3, namely 1α,25-dihydroxy-22-oxavitamin D3 (OCT) and 1α,25-dihydroxy-2β-(3-hydroxypropoxy) vitamin D3 (ED-71). OCT, a 22-oxa analog obtained from modification of the side chain of 1,25(OH)2D3, has been used clinically as an injection for the treatment of secondary hyperparathyroidism underlying renal insufficiency and as an ointment for the skin disease, psoriasis. OCT has also been reported to exhibit antiangiogenic activity, exerting antitumor effects without producing serious side effects such as hypercalcemia. On the other hand, ED-71, which possesses a hydroxypropoxy substituent at the 2β-position of the A-ring of 1,25(OH)2D3, has more potent biological effects on bone compared to 1 and phase III clinical studies for bone-fracture prevention have been completed. To explore structure activity relationship between ED-71 and related analogs, significant attention was now focused on the diastereomer of 3 at both the 1- and 3-positions of the A-ring, namely 3-epi-ED-71, 1-epi-ED-71 and 1,3-diepi-ED-71. All possible A-ring diastereomers at the 1- and 3-positions of ED-71 were synthesized using C2-symmetrical epoxide as a common starting material by convergent Trost methodology.
- 1α,25-Dihydroxyvitamin D3
- 1α,25-dihydroxy-2β-(3-hydroxypropoxy)vitamin D3
- ED-71
- 1-epi-ED-71
- 3-epi-ED-71
- 1,3-diepi-ED-71
Active vitamin D3, 1α,25-dihydroxyvitamin D3 (1,25 (OH)2D3, 1), is well recognized as a potent regulator of cell proliferation and differentiation in addition to possessing regulatory effects on calcium and phosphorus metabolism (1). Various analogs of 1,25(OH)2D3 (1) have been synthesized to separate differentiation-induction and antiproliferation activities from calcemic activity with the aim of obtaining useful analogs for the medical treatment of psoriasis, cancer, etc., without risk of hypercalcemia (2). 1α,25-Dihydroxy-22-oxavitamin D3 (OCT, 2), which contains an oxygen atom at the 22-position in the side chain of 1, was synthesized for this purpose. OCT has been shown to be highly potent in stimulating monocytic differentiation of human promyelocytic leukemic HL-60 cells but is less calcemic than 1. OCT has been used clinically as an injection for the treatment of secondary hyperparathyroidism underlying renal insufficiency and as an ointment for the skin disease, psoriasis (3). OCT has also been reported to exhibit antiangiogenic activity, exerting antitumor effects without producing serious side effects such as hypercalcemia. Furthermore, a number of in vitro and in vivo studies carried out in different animal species have demonstrated the antitumor properties of OCT for breast cancer, pancreatic cancer, prostate cancer, salivary cancer, lung cancer, etc. (4).
There is also intense interest in obtaining analogs more potent than 1 in regulating calcium and phosphorus metabolism with the objective of treating bone disease such as osteoporosis. 1α,25-Dihydroxy-2β-(3-hydroxypropoxy)vitamin D3 (ED-71, 3), which possesses a hydroxypropoxy substituent at the 2β-position of the A-ring of 1, is such an analog that shows more potent effects in bone therapy compared to 1. A phase III clinical trial with ED-71 has been completed with very good results as an oral medication for treating osteoporosis. The hope is that it will, in the near future, help osteoporosis patients as a characteristic new active vitamin D3 analog (Figure 1) (3, 5).
Recently, it has been reported that the epimerization of 1 at the 3-position of the A-ring plays a major role in parathyroid hormone (PTH) synthesis and secretion. Epimerized 3-epi-1,25(OH)2D3 shows equipotent and prolonged activities in comparison with 1 at suppressing PTH secretion (6, 7). During our clinical development of ED-71, the serum PTH in osteoporotic patients, however, was found not to change significantly upon treatment with ED-71 (5). A bulky hydroxylpropoxy substituent at the 2-position of the A-ring was assumed to interfere with epimerization of ED-71 at the adjacent and sterically hindered 3-position leading to the lack of epimerized 3-epi-ED-71 (4) in the parathyroid glands. This could explain why ED-71 showed weak potency in PTH suppression during clinical studies. Therefore, the synthesis and the biological evaluation of 4 were of interest. On the other hand, it has also been reported that the epimerization of 1 at the 1-position of the A-ring renders it devoid of activity as an agonist for transcaltachia concerning non-genomic intestinal calcium absorption (8). Therefore, 1-epi-1,25(OH)2D3 might be considered a potent stereospecific antagonist of 1 stimulating a transcaltachia response (8). Considering the structure-activity relationship between 1 and 1-epi-1,25(OH)2D3, the synthesis and biological activity of 1-epi-ED-71 (5) were also of interest. Moreover to further explore the structure-activity relationship between ED-71 and related analogs, significant attention was focused on the diastereomer of 3 at both the 1- and 3-positions of the A-ring, namely 1,3-diepi-ED-71 (6). In this paper, we describe the synthesis of all possible A-ring diastereomers at the 1- and 3-positions of ED-71, 4, 5 and 6, using C2-symmetrical epoxide (8) as a common starting material (Figure 2).
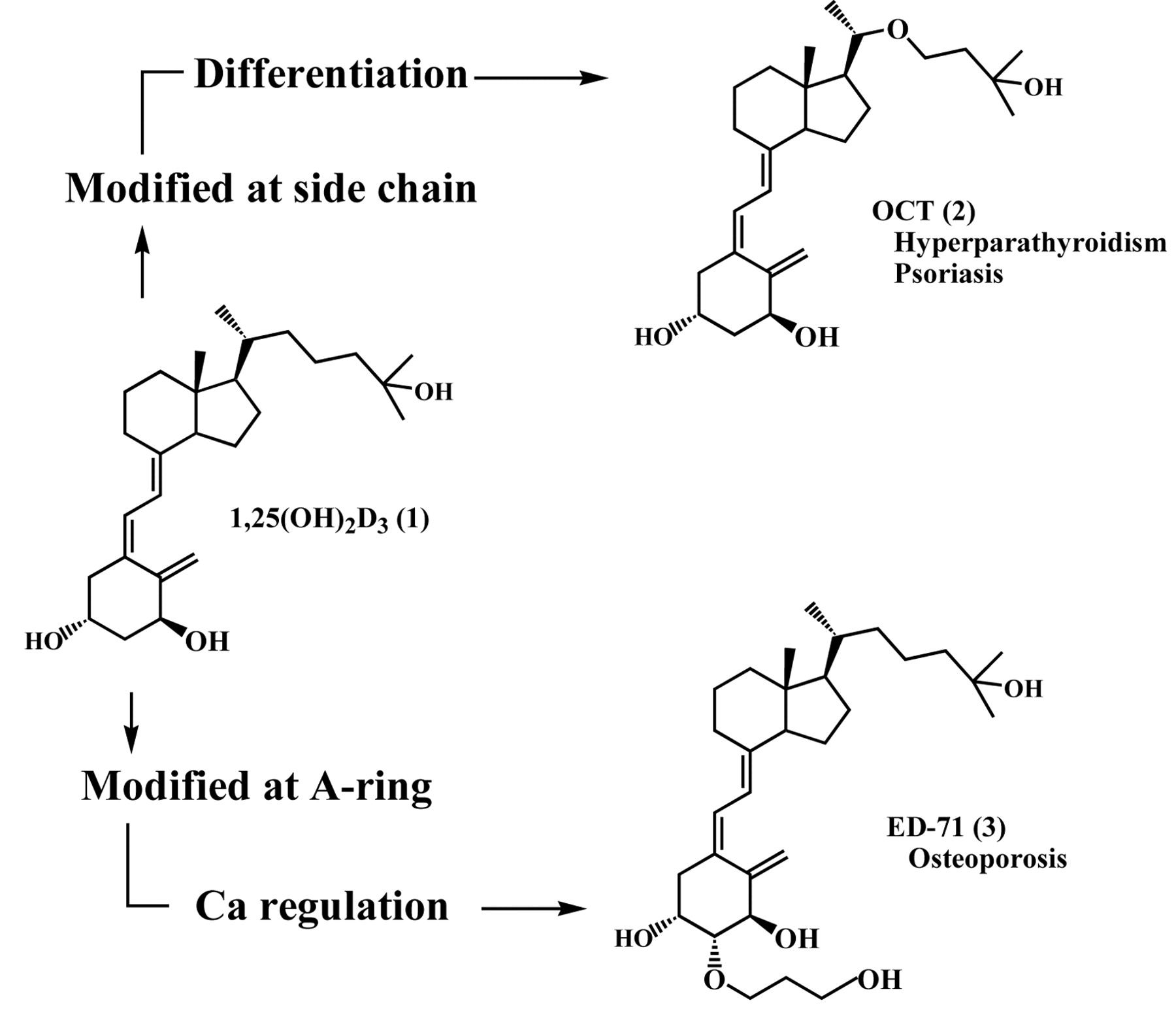
Modification of 1,25(OH)2D3 (1), chemical structure and therapeutic indication of OCT (2) and ED-71 (3).
Materials and Methods
General methods. All reactions were performed under argon atmosphere. All extracts were dried over magnesium sulfate and evaporated under reduced pressure with a rotary evaporator. Anhydrous tetrahydrofuran (THF) was purchased from Kanto Chemical Co. Inc., dichloromethane (CH2Cl2), triethylamine (Et3N), dimethylsulfoxide (DMSO), toluene, and acetonitrile (MeCN) were distilled from CaH2. Methanol (MeOH) was distilled from sodium. Thin-layer chromatography was performed with Merck F-254 TLC plates. Column chromatography was performed using Kanto Chemical Co. Inc., silica gel 60 N (spherical neutral). Infrared (IR) spectra were measured on a JASCO FTIR-230 spectrometer. Optical rotations were recorded on a JASCO DIP-370 polarimeter at ambient temperature. 1H NMR and 13C NMR spectra were measured on a Varian Gemini 300, JEOL JNM-AL 400, or Varian Unity plus 500 spectrometers. For 1H NMR spectra, chemical shifts are reported as δ values in ppm downfield from tetramethylsilane. For 13C NMR spectra, chemical shifts are reported as δ values in ppm relative to chloroform CHCl3 or MeOH. Mass spectra (MS) were measured with JEOL JMS-HX-100, Shimadzu GCMS QP-1000, and Hitachi M1200H instruments. High-resolution mass spectra (HRMS) were recorded on JEOR JMS-AX-500 and VG Auto Spec Q instruments. Ultraviolet (UV) spectra obtained with Shimadzu UV-240 spectrometer using ethanol (EtOH) as a solvent.
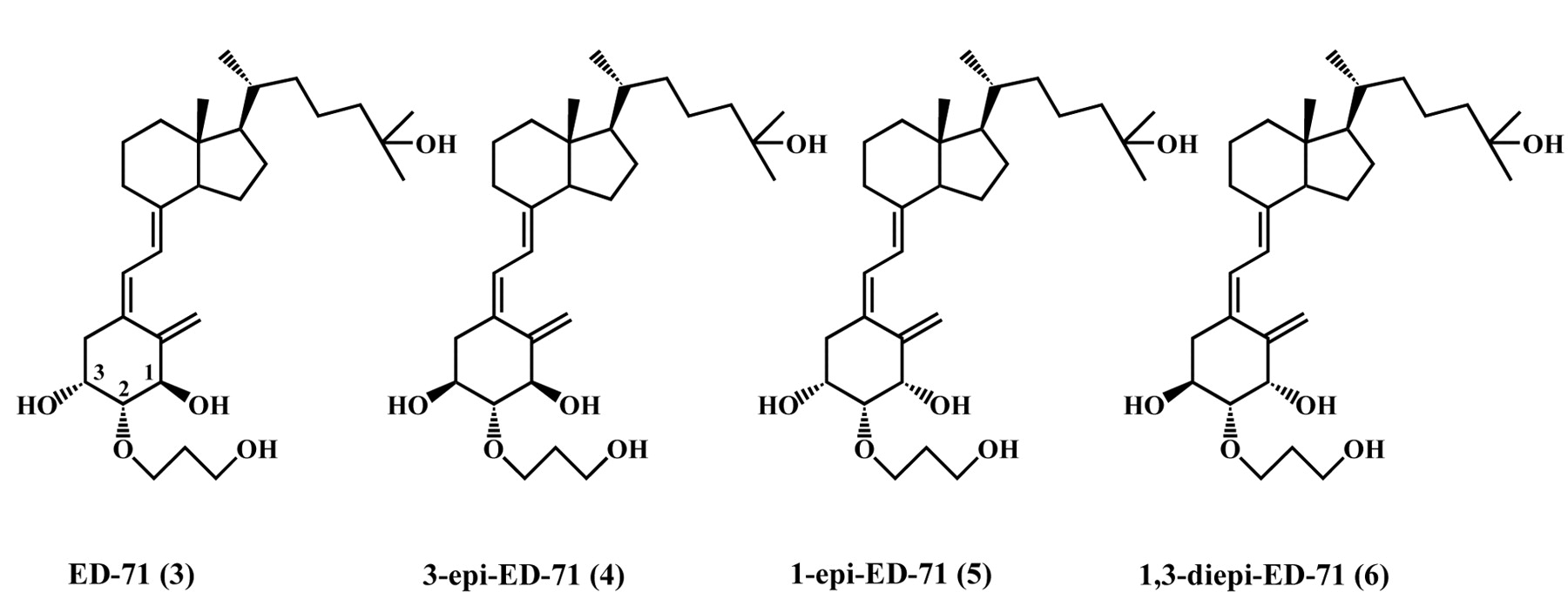
Structure of ED-71 (3), 3-epi-ED-71 (4), 1-epi-ED-71 (5) and 1,3-diepi-ED-71 (6).
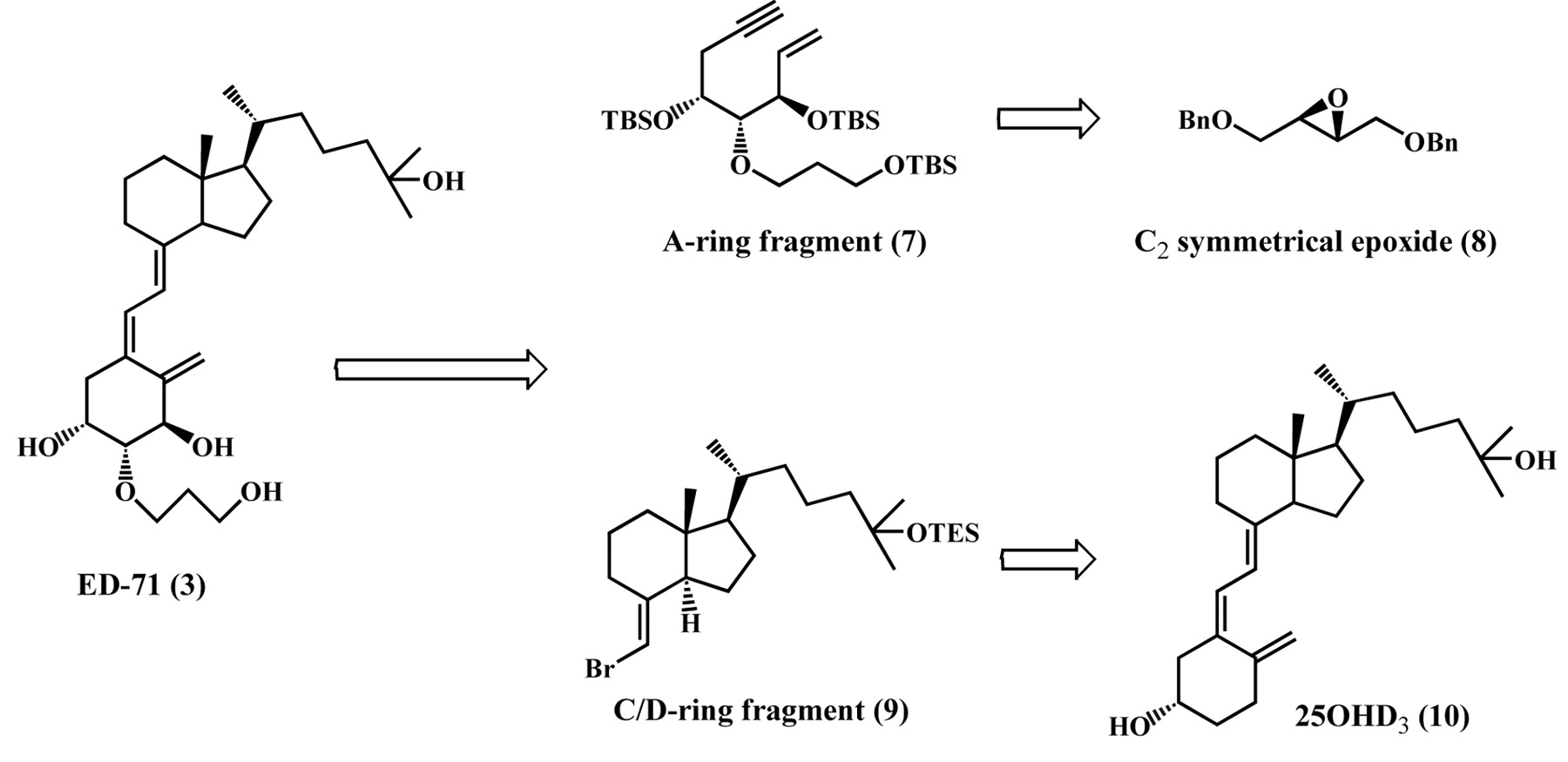
Retrosynthesis of ED-71 (3).
(5Z,7E)-(1R,2R,3R)-2-(3-Hydroxypropoxy)-9,10-secocholesta-5,7,10(19)-triene-1,3,25-triol (3). ED-71 (3) was prepared from A-ring fragment (7) and C/D-ring fragment (9) as a colorless foam; IR (nujol): ν 3360, 1100, 1060, 910 cm-1. 1H NMR (CDCl3): δ 6.36 (1H, d, J=10.5 Hz), 6.04 (1H, d, J=10.5 Hz), 5.49 (1H, s), 5.08 (1H, s), 4.36-4.12 (2H, m), 4.02-3.60 (5H, br), 1.21 (6H, s), 0.91 (3H, d, J=6.1 Hz), 0.55 (3H, s). MS (EI) m/z 490 (M+), 472, 454, 396, 59 (100%). HRMS (EI) calcd for C30H50O5 (M+) 490.3658, found 490.3678. UV λmax; 263 nm.
(5Z,7E)-(1S,2R,3R)-2-(3-Hydroxypropoxy)-9,10-secocholesta-5,7,10(19)-triene-1,3,25-triol (4). 1-Epi-ED-71 (4) was prepared from A-ring fragment (17) and C/D-ring fragment (9) as a white powder; IR (neat): ν 3400, 2930, 2850 cm-1. 1H NMR (CDCl3): δ 6.43 (1H, d, J=12.0 Hz), 6.03 (1H, d, J=12.0 Hz), 5.38 (1H, s), 5.08 (1H, s), 4.33-4.28 (1H, br s), 4.10-4.01 (1H, m), 3.91 (2H, t, J=5.4 Hz), 3.84 (2H, t, J=5.4 Hz), 3.64-3.58 (1H, m), 1.22 (6H, s), 0.94 (3H, d, J=6.5 Hz), 0.54 (3H, s). HRMS (EI) calcd for C30H50O5 (M+) 490.3658, found 490.3706. UV λmax: 264 nm, λmin: 227 nm.

Synthesis of ED-71 (3). Reagents and conditions: a) HO(CH2)3OH/t-BuOK, 120°C. b) t-BuCOCl/pyridine/CH2Cl2, rt. c) i) H2/Pd(OH)2/MeOH, rt. ii) Me2C(OMe)2/TsOH/acetone, rt. d) i) DMSO/(COCl)2/CH2Cl2, -60°C. ii) CH2=CHMgBr/THF, -60°C. iii) t-BuCOCl/Et3N/DMAP/CH2Cl2, rt. e) 1 M HCl/MeOH, rt. f) Ph3P/DEAD/benzene, reflux. g) i) LiC CTMS/BF3-OEt2, -78°C. ii) 10 N NaOH/MeOH, rt. iii) TBSOTf/Et3N/CH2Cl2, 0°C. h) i) TESOTf/Et3N/CH2Cl2, 0°C. ii) O3/CH2Cl2/MeOH, -78°C then NaBH4/MeOH, -78°C. i) NMO/TPAP/4Ams/CH2Cl2, rt. j) Ph3P+CH2BrBr-/NaHMDS/THF, -60°C - rt. k) (dba)3Pd2-CHCl3/PPh3/Et3N/toluene, reflux. l)TBAF/THF/toluene, reflux.
(5Z,7E)-(1R,2R,3S)-2-(3-Hydroxypropoxy)-9,10-secocholesta-5,7,10 (19)-triene-1,3,25-triol (5). 3-Epi-ED-71 (5) was prepared from A-ring fragment (30) and C/D-ring fragment (9) as colorless crystals; mp 126-128°. [α]D26 -61.6° (c, 0.39, CH3OH). IR (KBr): ν 3332, 2939, 1641, 1442, 1375, 1082 cm-1. 1H NMR (CD3OD) δ 6.32 (1H, d, J=9.8 Hz), 5.99 (1H, d, J=11.2 Hz), 5.13 (2H, dt, J=2.4, 24.3 Hz), 3.96-3.87 (2H, m), 3.79 (1H, dt, J=2.2, 8.9 Hz), 3.71 (2H, t, J=6.1 Hz), 3.51-3.46 (1H, m), 2.97 (1H, t, J=8.9 Hz), 2.84 (1H, dd, J=5.0, 11.2 Hz), 2.50 (1H, dd, J=5.2, 12.8 Hz), 2.17 (1H, t, J=11.1), 2.04-1.99 (2H, m), 1.99-1.87 (1H, m), 1.82 (2H, quint, J=6.1 Hz), 1.70-1.66 (2H, m), 1.58-1.40 (7H, m), 1.37-1.28 (4H, m), 1.26-1.22 (1H, m), 1.16 (6H, s), 1.10-1.03 (1H, m), 0.96 (3H, d, J=6.4 Hz), 0.58 (3H, s). 13C NMR (CD3OH) δ 147.3, 143.6, 134.0, 124.5, 118.7, 111.8, 90.2, 75.3, 73.0, 71.5, 71.4, 60.5, 58.0, 57.6, 47.1, 45.3, 43.6, 41.8, 37.7, 33.7, 30.0, 29.3, 29.1, 28.7, 24.8, 23.4, 21.9, 19.4, 12.3. HRMS (EI) m/z calcd for C30H50O5 (M+) 490.3658, found 490.3658.
(5Z,7E)-(1S,2R,3S)-2-(3-Hydroxypropoxy)-9,10-secocholesta-5,7,10(19)-triene-1,3,25-triol (6). 1,3-Diepi-ED-71 (6) was prepared from A-ring fragment (36) and C/D-ring fragment (9) as a colorless oil. [α]D22 -18.3° (c 0.12, MeOH). IR (neat): ν 3359, 2941, 1375, 1076 cm-1. 1H NMR (CD3OD): δ 6.25 (1H, d, J=11.2 Hz), 5.99 (1H, d, J=11.2 Hz), 5.24 (1H, s), 4.88 (1H, s), 4.32 (1H, d, J=3.0 Hz), 3.88-3.87 (1H, m), 3.70-3.67 (1H, m), 3.60-3.56 (3H, m), 2.76 (1H, dd, J=3.4, 11.2 Hz), 2.51 (1H, dd, J=4.9, 13.6 Hz), 2.07 (1H, dd, J=6.8, 13.2 Hz), 1.95-1.87 (2H, m), 1.82-1.79 (1H, m), 1.72 (2H, quint, J=5.8 Hz), 1.58 (2H, d, J=11.2 Hz), 1.41-1.31 (7H, m), 1.26-1.19 (4H, m), 1.07 (6H, s), 0.87 (3H, d, J=6.8 Hz), 0.80-0.77 (1H, m), 0.47 (3H, s). 13C NMR (CD3OD): δ 147.1, 134.6, 125.2, 119.0, 115.0, 85.8, 73.0, 71.5, 69.4, 68.6, 60.4, 58.0, 57.6, 45.3, 42.4, 41.9, 37.8, 33.7, 30.8, 30.0, 29.3, 29.1, 28.7, 24.7, 23.4, 21.9, 19.4, 12.3. HRMS (FAB) calcd for C30H50O5 (M+) 490.3658 found 490.3648.
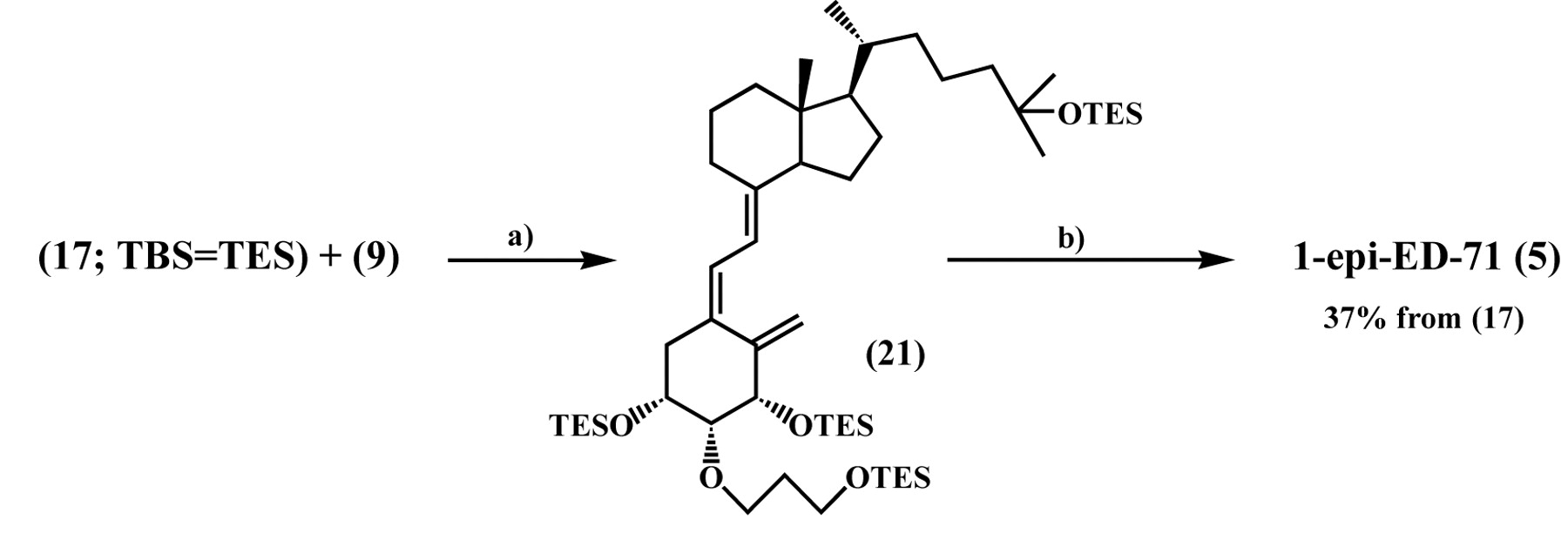
Synthesis of 1-epi-ED-71 (5). Reagents and conditions: a) Pd(PPh3)4/Et3N/toluene, reflux. b) 47% HF/MeCN, rt.
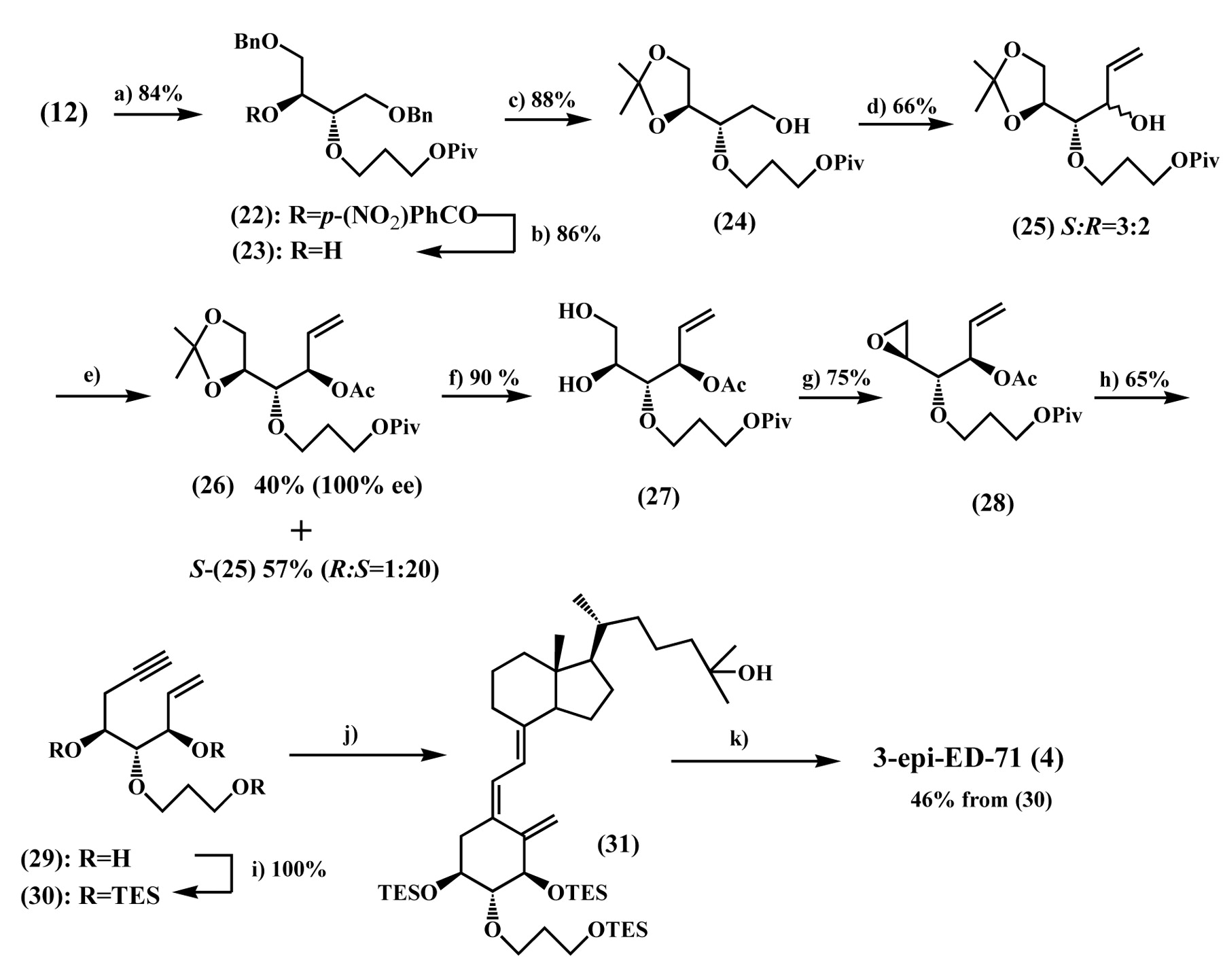
Synthesis of 3-epi-ED-71 (4). Reagents and conditions: a) p-(NO2)PhCO2H/DEAD/PPh3/toluene, rt. b) NaHCO3/MeOH, rt. c) i) Pd(OH)2/H2/MeOH, rt. ii) 2,2-dimethoxypropane/TsOH/acetone, rt. d) i) (COCl)2/DMSO/CH2Cl2, -78°C then Et3N. ii) CH2=CHMgBr/THF, -40°C. e) Novozyme/CH2=CHOAc/t-BuOMe, 30°C. f) 60% AcOH/H2O, rt. g) DEAD/PPh3/dioxane, reflux. h) i) LiC=CTMS/BF3-OEt2, -78°C. ii) 10 M NaOH/MeOH, rt. i) TESOTf/Et3N/CH2Cl2, -40°C. j) (9; TES=H)/Pd(PPh3)4/Et3N/toluene, reflux. k) NH4F/MeOH, reflux.
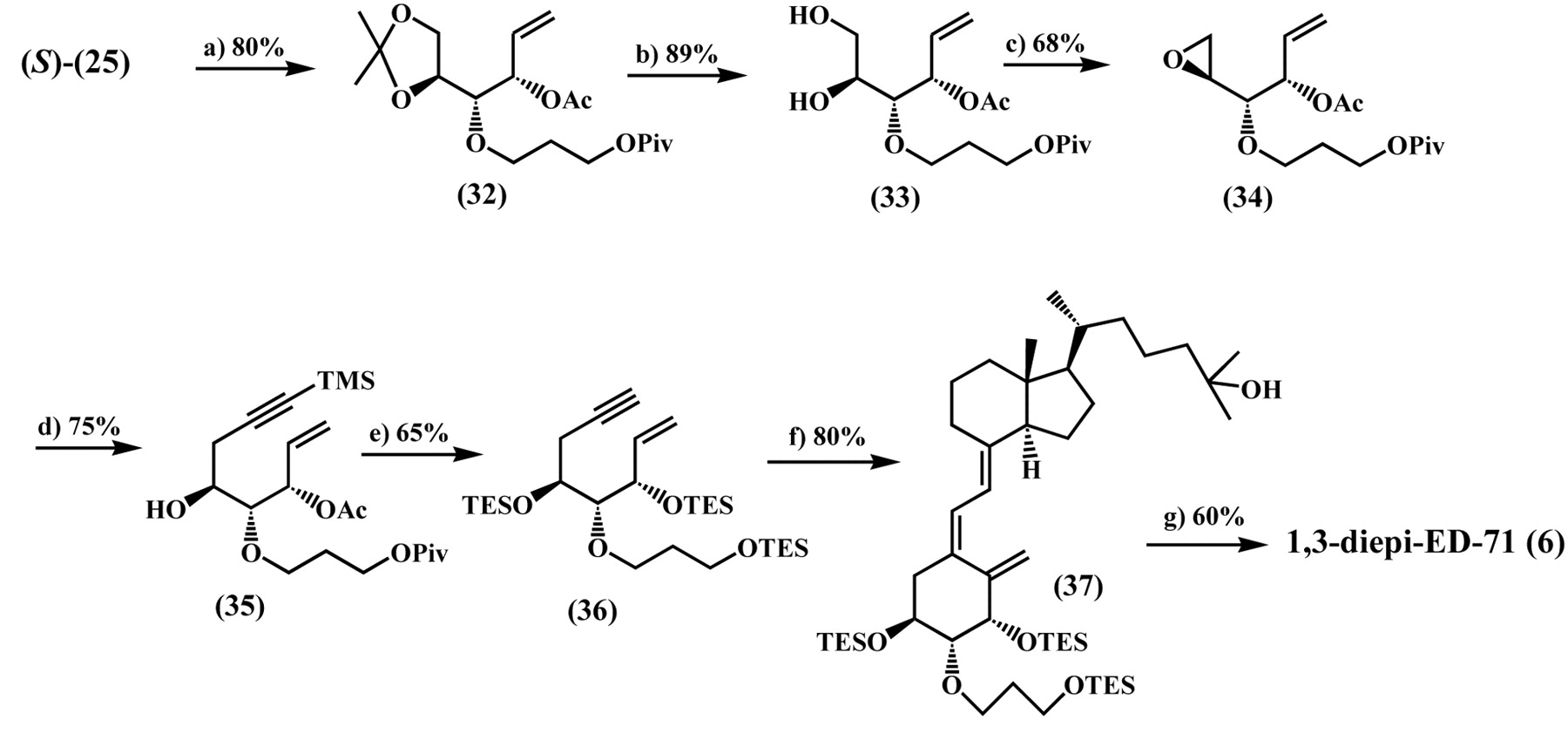
Synthesis of 1,3-diepi-ED-71 (6). Reagents and conditions: a) Ac2O/Et3N/DMAP/CH2Cl2, rt. b) 60% AcOH/H2O, rt. c) PPh3/DEAD/dioxane, reflux. d) LiC=CTMS/BF3-OEt2/THF, -78°C. e) i) 10 M NaOH/MeOH, rt. ii) TESOTf/Et3N/CH2Cl2, -40°C. f) (9; TES=H)/Pd(PPh3)4/Et3N/toluene, reflux. g) 46% HF/MeCN, rt.
Results and Discussion
The synthesis of all the possible A-ring diastereomers at the 1- and 3-positions of ED-71 (3) was envisioned using convergent methodology described by Trost et al. (9, 10). The key step involves palladium-catalyzed coupling of A-ring fragment (7) prepared from C2-symmetrical epoxide (8) with C/D-ring fragment (9) obtained from 25-hydroxyvitamin D3 (25OHD3, 10). This methodology was evaluated as an improved method for industrial scale production of ED-71, considering the potential clinical application of 3 as a useful drug in the near future (Figure 3) (11).
Synthesis of ED-71(3) and 1-epi-ED-71 (5). The required A-ring fragment (7) for the synthesis of ED-71 (3) was synthesized based on the methodology which we have previously established (11). Thus, cleavage of the known C2-symmetrical epoxide (8) (12) with 1,3-propanediol in the presence of potassium tert-butoxide (t-BuOK) gave the diol (11) in 86% yield. After protection of the primary hydroxyl group to give the pivalate (12) in 88% yield, cleavage of the benzyl ether moiety in 12 and subsequent protection of the resulting 1,2-diol as the acetonide gave the alcohol (13) in 87% overall yield. Swern oxidation of 13 and subsequent Grignard reaction of the resulting aldehyde with vinylmagnesium bromide (CH2=CHMgBr) followed by pivaloylation of the resulting alcohol afforded the dipivalate (14) as an epimeric mixture (R/S=3/2). Without separation of the epimeric mixture, the acetonide moiety in 14 was cleaved quantitatively to give the diol (15). Exposure of 15 to Mitsunobu conditions (13) afforded the epimeric epoxide (16) in 77% yield. The acetylene unit was successfully installed by the regioselective epoxide-opening of 16 with lithium trimethylsilylacetylide (LiC=CTMS) to provide the ene-yne (7) as the A-ring fragment for ED-71 (3) in 36% yield after protecting group exchange from the pivalate to the tert-butyldimethylsilyl (TBS) ether. The accompanying (S)-epimer (17), which consists of the requisite stereochemistry to obtain 1-epi-ED71 (6), was separated in 24% yield by simple column chromatography. Next, the synthesis of the C/D-ring fragment (9) from readily and commercially available 25OHD3 (10) was performed (14). 25OHD3 (10) was protected as the bis-triethylsilyl (TES) ether using triethylsily trifluoromethanesulfonate (TESOTf), and was then converted to the alcohol (18) by ozonolysis and treatment with sodium borohydride (NaBH4) (72% yield from 10). The hydroxyl moiety in 18 was oxidized to the ketone (19) with tetrapropylammonium perruthenate (TPAP) and N-methyl-molpholine N-oxide (NMO) in 99% yield. Wittig reaction of 19 with (bromomethylene) triphenyl-phosphonium bromide (Ph3P+CH2Br/Br-) and sodium hexamethyldisilazide (NaHMDS) gave rise to the C/D-ring fragment bromomethylene (9) in 38% yield. With A-ring fragment (7) and C/D-ring fragment (9) in hand, next the Trost coupling reaction was investigated. Thus, upon treatment of 7 and 9 with Et3N, triphenyphosphine (PPh3) and tris (dibenzylidene-acetone)dipalladium-chloroform [(dba)3Pd2-CHCl3] in boiling toluene, the coupled product (20) was obtained in 26% yield together with recovered 7 (45%) and 9 (56%). Deprotection of the silyl moiety in 20 with tetrabutylammonium fluoride (TBAF) afforded ED-71 (3) in 60% yield (Figure 4) (14).
On the other hand, as described above, when the ene-yne (7) was prepared from the epimeric epoxide (16) as the (R)-isomer, the separable (S)-isomer (17) was accompanied as a by-product, which was used to obtain 1-epi-ED-71 (5). Upon treatment of excess bromomethylene (9) and ene-yne (17, TBS=TES) in the presence of tetrakis (triphenylphosphine)-palladium (0) [Pd(PPh3)4] and Et3N in boiling toluene, the coupled product (21) was obtained as an inseparable mixture with recovered 9. The mixture was desilylated using 47% hydrofluoric acid (HF) in MeCN and purified to afford 1-epi-ED-71 (5) in 37% yield from 17 (Figure 5) (15, 16).
Synthesis of 3-epi-ED-71 (4) and 1,3-diepi-ED-71 (6). The synthesis of the A-ring fragment (30) for 3-epi-ED-71 (4) began with inversion of the C3 configuration of the alcohol (12). Reaction of 12 with p-nitrobenzoic acid in the presence of diethyl azodicarboxylate (DEAD) and PPh3 gave p-nitrobenzoate (22) in 84% yield (17). Treatment of 22 with sodium bicarbonate (NaHCO3) in MeOH allowed selective methanolysis of the p-nitrobenzoate group to give the inverted alcohol (23) in 86% yield. After hydrogenolysis of the benzyl ether functionalities in 23 the resulting diol was protected as its acetonide to afford the acetonide (24) in 88% yield. Swern oxidation of 24 followed by Grignard reaction of the resulting aldehyde with CH2=CHMgBr produced the alcohol (25) as an epimeric mixture (S:R=3:2) in 66% yield. To separate this epimeric mixture, 25 was subjected to lipase-catalyzed acetylation using vinyl acetate (CH2=CHOAc) and Novozyme in t-butyl methyl ether (t-BuOMe) (18). As a result, the R-epimer preferentially underwent acetylation to give the acetate (26) and S-25 (R:S=1:20) in 40% and 57% yields, respectively. Acidic hydrolysis of 26 gave the diol (27) in 90% yield, which upon Mitsunobu reaction using DEAD and PPh3 in boiling toluene afforded the epoxide (28) in 75% yield (12). Reaction of 28 with LiC=CTMS in the presence of boron trifluoride diethyl etherate (BF3·OEt2) at -78°C followed by saponification provided the ene-yne (29) in 65% yield (19). Protection of 29 as its TES ether produced the A-ring fragment (30) quantitatively. Having secured the A-ring fragment (30), its coupling with the C/D-ring fragment (9) was performed using the Trost methodology. Thus, the A-ring fragment (30) was allowed to react with the C/D-ring fragment (9, TES=H) in the presence of Pd(Ph3P)4 and Et3N in boiling toluene to give the coupling product (31) which was desilylated with ammonium fluoride (NH4F) in boiling MeOH to produce 3-epi-ED-71 (4) in 46% yield from 30 (Figure 6) (20).
The synthesis of the A-ring fragment (36) for 1,3-diepi-ED-71 (6) started from the alcohol (25) which was obtained in our previous lipase-catalyzed acetylation of 25 as the unreacted (S)-isomer. The alcohol (25) possesses the requisite stereochemistry at positions 1, 2 and 3 of the A-ring that comprises 6. Acetylation of 25 gave the acetate (32) in 80% yield from which the diol (33) was obtained in 89% yield after deprotection of the acetonide moiety using 60% acetic acid (AcOH). Mitsunobu reaction of 33 with DEAD and PPh3 in boiling toluene afforded the epoxide (34) in 68% yield. Reaction of 34 with LiC=CTMS in the presence of BF3·OEt2 at -78°C gave the ene-yne (35) in 75% yield, which was then converted to the A-ring fragment (36) by saponification with 10 M sodium hydroxide (NaOH) and subsequent protection of the hydroxyl groups as their TES ether in 65% overall yield. Next the A-ring fragment (36) was coupled to the C/D-ring fragment (9) using Trost's methodology. Thus, 36 was coupled with 9 (TES=H) in the presence of Pd(PPh3)4 and Et3N in boiling toluene to produce the desired coupling product (37) in 80% yield. Desilylation of 37 with 46% HF in MeCN at room temperature gave rise to 1,3-diepi-ED-71 (6) in 60% yield (Figure 7) (21).
Conclusion
Based on the Trost coupling methodology involving A-ring fragments (7), (17), (30) and (36) and C/D-ring fragment (9), the synthesis of ED-71 (3), 3-epi-ED-71 (4), 1-epi-ED-71 (5) and 1,3-diepi-ED-71 (6) was successfully accomplished, completing the preparation of the full complement of A-ring 1-and 3-positional diasteromers. The detailed biological properties of these analogs are currently under investigation and will be reported elsewhere.
Acknowledgements
We are grateful to Professor David Horne of the Division of Molecular Medicine, City of Hope for helpful suggestions and reading of the manuscript.
- Received January 29, 2009.
- Revision received April 2, 2009.
- Accepted May 21, 2009.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}