


Abstract
Background: The tumor suppressor gene TMS1 (target of methylation-induced silencing) has been described in the literature as a pro-apoptotic gene. This study examined the methylation status of TMS1 in breast cancer cells and its potential role in sensitivity to docetaxel chemotherapy. Materials and Methods: Methylation of the TMS1 promoter was examined by methylation-specific PCR (MS-PCR) and gene expression was analyzed by reverse transcriptase PCR (RT-PCR). Apoptosis was evaluated by annexin V/propidium iodide staining followed by flow cytometric analysis. Results and Conclusion: The TMS1 promoter was unmethylated in ZR75-1, MB-231 and MCF7 cells which expressed the gene and partially methylated in SKBR3 and Hs578t cells in which TMS1 expression was down-regulated. Treatment of SKBR3 and Hs578t cells with demethylating agents resulted in reactivation of the TMS1 gene. Pretreatment with 5-azacytidine increased sensitivity to docetaxel treatment in SKBR3 and Hs578t cells, indicating that TMS1 reactivation in these cells may contribute to docetaxel sensitivity.
Apoptosis or programmed cell death occurs as part of normal cellular processes including maintenance of cell homeostasis and also as a response to exposure to ultraviolet radiation or DNA damaging agents such as chemotherapeutic drugs. In cancer cells, defects in the apoptotic pathway contribute to resistance to chemotherapy (1). Epigenetic silencing of tumor suppressor and pro-apoptotic genes is one of the mechanisms of development of resistance to anticancer drugs (1, 2).
TMS1 (target of methylation induced silencing), also known as ASC (apoptosis speck-like protein containing a CARD) has been described in the literature as a gene which encodes a protein containing a pyrin domain (PYD) in the N-terminus and a caspase recruitment domain (CARD) in the C-terminus. Both PYD and CARD are members of the death domain-fold superfamily of proteins and are involved in the apoptotic and inflammatory signaling pathways (3). Previous studies have shown that the overexpression of TMS1 induced caspase 9-dependent apoptosis (4). Futhermore, the knockdown of TMS1 reduced sensitivity of cells to cytotoxic agents (5). This indicated the critical role of TMS1 in apoptosis. Earlier reports have shown that TMS1 is silenced by DNA hypermethylation in several carcinomas including that of the prostate, ovary and breast (6-9).
This study examined the effect of promoter methylation on the regulation of TMS1 in breast cancer and its potential role in docetaxel chemotherapy, a currently used treatment for breast cancer.
Materials and Methods
Cell culture. Breast cancer cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). MCF7, Hs578t, ZR75-1 and SKBR3 cells were grown in RPMI-1640 and MB-231 in DMEM-F12 supplemented with 10% fetal bovine serum, 2 mM glutamine (Invitrogen, Carlsbad, CA, USA) and 100 μg/ml penicillin-streptomycin (Invitrogen) in a humidified incubator at 37°C with 5% CO2.
Drug treatment. 5-Azacytidine and 5-azadeoxycytidine were purchased from Sigma Aldrich (St. Louis, MO, USA). The SKBR3 and Hs578t cells were treated with 5-azacytidine or 5-azadeoxycytidine for 72 h, after which RNA was extracted. For combination treatment, the SKBR3 and Hs578t cells were seeded in 12-well plates, and treated with 5-azacytidine for 72 h followed by 2.5 nM or 5 nM docetaxel for 72 h.
RNA extraction and reverse transcriptase (RT) PCR. RNA was extracted using RNA Stat 60 (Tel-test, Friendswood, TX, USA), as per the manufacturer's instructions. A total of 2.5 μg of RNA was reverse transcribed in a 25 μl reaction containing 1× M-MLV reaction buffer, 1.25 μl of dNTP mix, 2.5 μl of random hexamers (GE Healthcare, Piscataway, NJ, USA) and 250 U of M-MLV Reverse Transcriptase (USB Corporation, Cleveland, OH, USA). PCR amplification was carried out using Jumpstart Red taq Ready mix (Sigma, St Louis, MO, USA) using the primers 5′GGA CGC CTT GGC CCT CAC CG 3′(forward) and 5′ GGC GCG GCT CCA GAG CCC TG 3′ (reverse) as previously described (6).

Methylation analysis of TMS1 in breast cancer cell lines. Genomic DNA was extracted from breast cancer cells, bisulfite treated and analyzed by MS-PCR using primers specific for unmethylated (U) and methylated (M) sequences. Water was used as negative control (NC).
Methylation-specific PCR (MS-PCR). DNA from the breast cancer cell lines was extracted using DNA Stat 60 (Tel-test). The genomic DNA was bisulfite treated as previously described (10). MS-PCR was performed using primers specific for methylated or unmethylated sequences in the TMS1 promoter region as previously described by Virmani et al. (11).
Apoptosis assay. Apoptosis after drug treatment was measured by annexin V/propidium iodide staining using an Annexin-V-FLUOS staining detection kit (Roche, Indianapolis IN, USA) followed by flow cytometry in a Beckman Coulter XL flow cytometer (Beckman Coulter Inc, Miami, FL, USA). Experiments were performed in triplicates and repeated 3 times for validation.
Results
Methylation status of TMS1 promoter in breast cancer cells. Epigenetic silencing of pro-apoptotic genes is one of the mechanisms of resistance of cancer cells to chemotherapy (1, 2). Previous studies have described the role of TMS1, containing both PYD and CARD domain, in apoptosis (4). The methylation status of the TMS1 promoter region was examined by MS-PCR using primers for unmethylated and methylated sequences as previously described (6, 11). As shown in Figure 1, we found that the proximal promoter region of TMS1 is unmethylated in ZR75-1, MCF7 and MB-231 and partially methylated in Hs578t and SKBR3.
TMS1 expression in breast cancer cells. The mRNA levels of TMS1 in the breast cancer cell lines were analyzed by RT PCR. As seen in Figure 2, high levels of TMS1 expression were seen in the ZR75-1, MB-231 and MCF7 cells in which the proximal promoter region of the gene was found to be unmethylated. TMS1 was down-regulated in the Hs578t and SKBR3 cells which showed partial methylation of the promoter region. These results indicate that down-regulation of TMS1 expression was associated with partial methylation of the promoter in the Hs578t and SKBR3 cells.
Effect of demethylating agents on TMS1 expression. As described above, TMS1 promoter is partially methylated in Hs578t and SKBR3, which corresponds to down-regulation of gene expression. In order to confirm that the deregulation of TMS1 in the Hs578t and SKBR3 cells was attributed to DNA methylation of the promoter, the cells were treated with the demethylating agents, 5-azacytidine and 5-azadeoxycytidine. Figure 3A and 3B shows the reactivation of TMS1 in the SKBR3 and Hs578t cells.
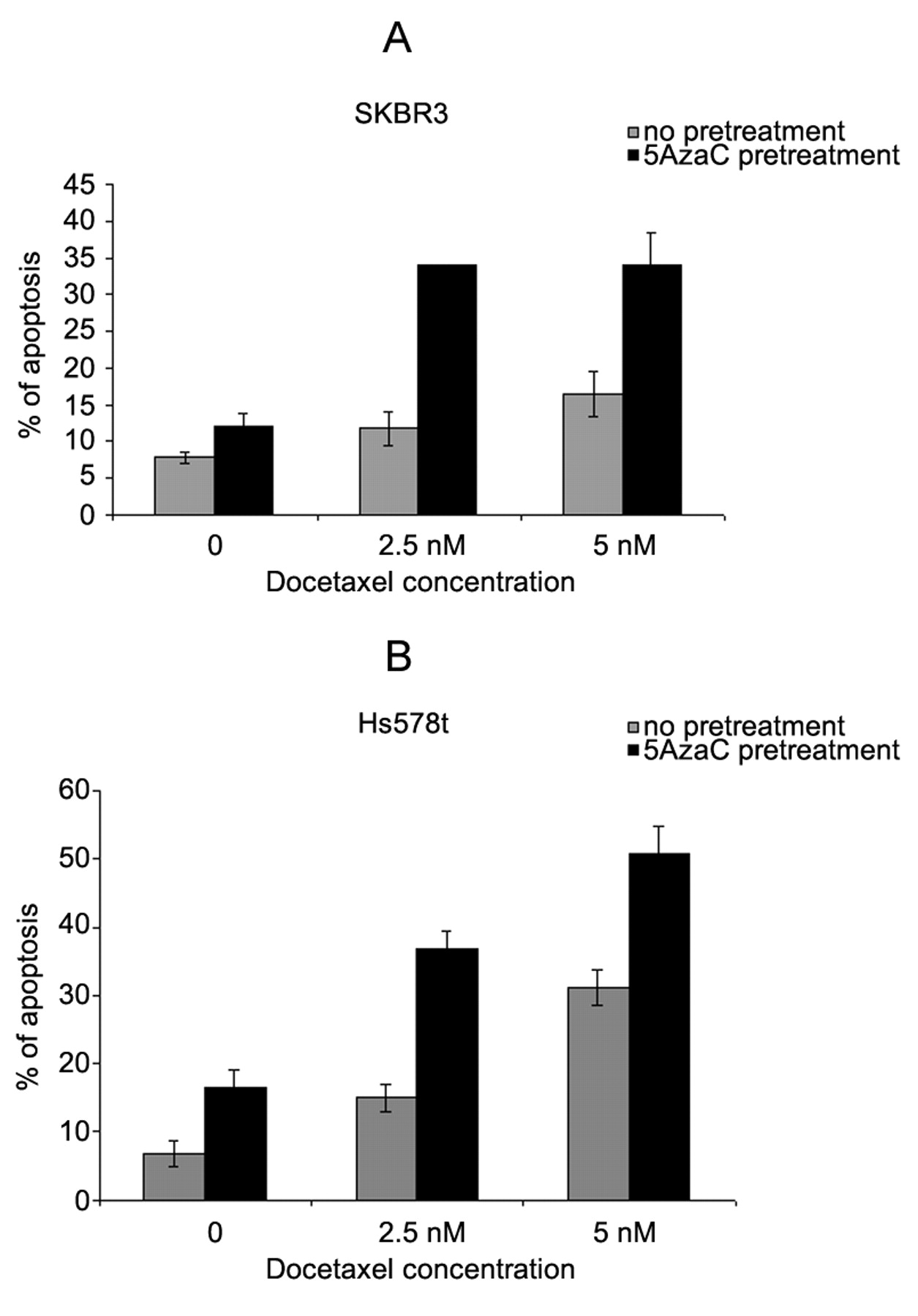
Effect of pretreatment with 5-azacytidine on sensitivity to docetaxel. The role of TMS1 in apoptosis has been documented in the literature (5, 8). Overexpression of TMS1 in breast cancer cells resulted in apoptosis and inhibition of cell growth (8). Our results showed that treatment of SKBR3 and Hs578t with demethylating agents reactivated TMS1 expression. Hence, we hypothesized that pretreatment of SKBR3 and Hs578t cells with 5-azacytidine would result in enhanced sensitivity to a currently used treatment for breast cancer, docetaxel chemotherapy. Based on dose titration of 5-azacytidine and docetaxel in these cells, for combination treatment, the minimum cytotoxic dose of 1.25 μM that resulted in re-expression of TMS1 (data not shown) was selected. The SKBR3 and Hs578t cells were treated with 1.25 μM 5-azacytidine for 72 h followed by treatment with 2.5 nM or 5 nM docetaxel for 72 h. As shown in Figure 4, the SKBR3 cells pretreated with 5-azacytidine showed increased sensitivity to docetaxel chemotherapy as compared to the cells treated with docetaxel alone. Similar results were also observed in the Hs578t cells. These data show that pretreatment of cells with demethylating agents such as 5-azacytidine up-regulates TMS1, thereby increasing sensitivity to docetaxel chemotherapy.
Discussion
The partial methylation of the TMS1 promoter in Hs578t and SKBR3 breast cancer cells correlated with the down-regulation of TMS1 expression in these cells. Treatment of the Hs578t and SKBR3 cells with DNA methyl transferase inhibitors (5-azacytidine and 5-azadeoxycytidine) resulted in up-regulation of TMS1 expression indicating that the down-regulation of TMS1 expression in SKBR3 and Hs578t is regulated by methylation of the promoter. Pretreatment with 5-azacytidine enhanced sensitivity to docetaxel treatment suggesting a probable role of TMS1 in docetaxel-mediated apoptosis in these cells.

Analysis of TMS1 expression in breast cancer cells. RNA extracted from breast cancer cells was reverse transcribed and amplified by PCR. RNA integrity was analyzed by β-actin amplification. Water was used as negative control (NC).
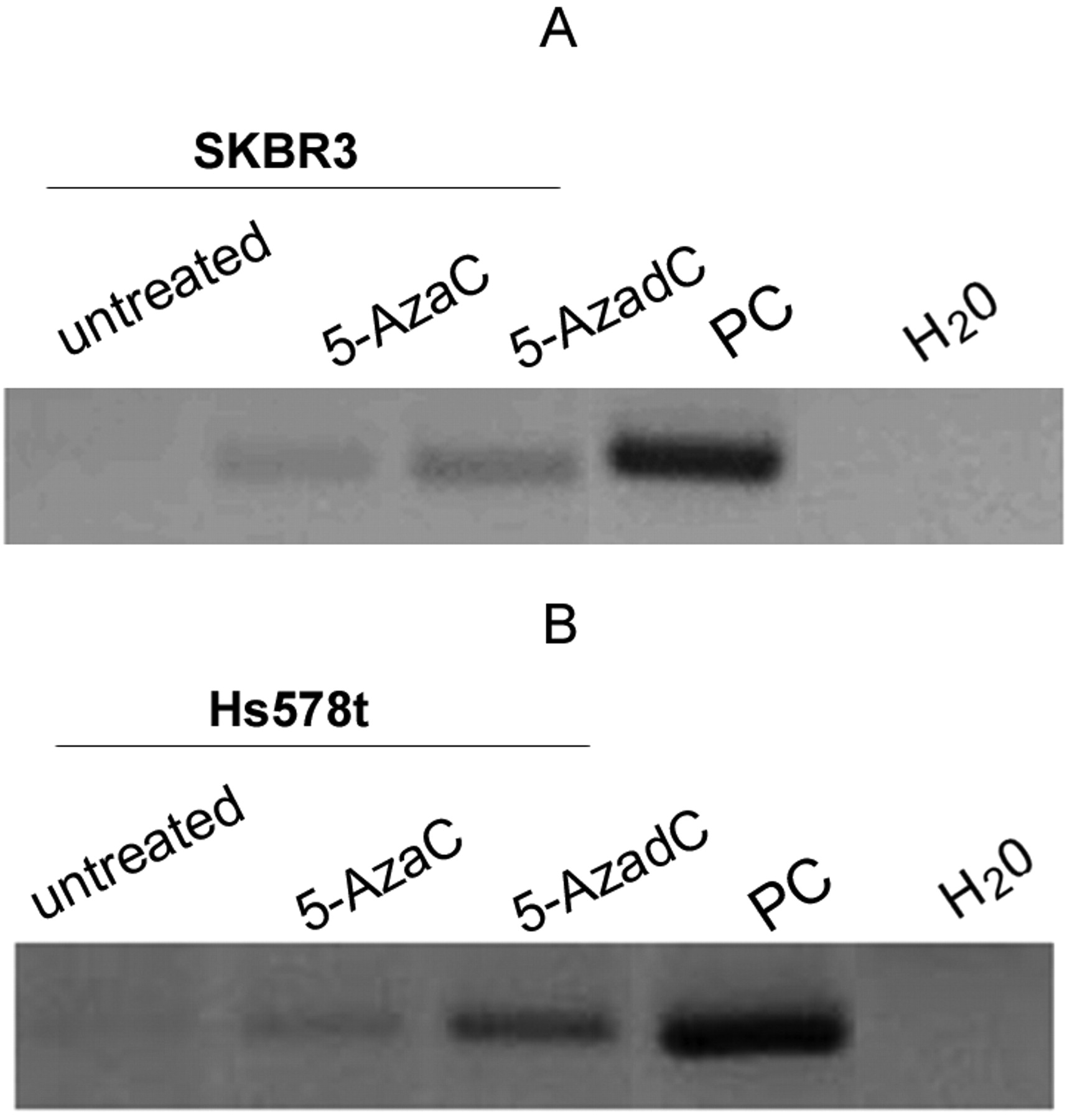
Reactivation of TMS1 in SKBR3 and Hs578t cells treated with DNMT inhibitors. RT-PCR analysis of (A) SKBR3 cells and (B) Hs578t cells treated with 1.25 μM 5-azacytidine (5-AzaC) or 5-azadeoxycytidine (5-AzadC) for 72 h. RNA from MCF7 was used as positive control (PC) and water was used as negative control (NC). RNA integrity was analyzed by β- actin amplification.
The methylation of CpG islands in the promoter region results in down-regulation or silencing of genes and is known to contribute to carcinogenesis, tumor progression, and resistance to apoptosis (12). DNA methylation modifications occur without changing the actual sequence however these changes can affect gene expression. Epigenetic silencing can occur as a result of blocking of transcription factor binding to the methylated sequence. Alternatively gene silencing can occur by the association of methyl binding domain proteins with the methylated CpGs resulting in alteration of the chromatin structure (13). Typically complete methylation of the proximal promoter region results in silencing of gene expression and partial methylation of CpG dinucleotides results in active gene expression. In contrast to this, the partial methylation of the TMS1 promoter in the present study was associated with gene silencing in the SKBR3 and Hs578t cells. Previously Conway et al. observed similar results on TMS1 methylation and expression in SKBR3 cells and it was suggested that methylation levels in these cells are above a critical threshold, resulting in gene silencing, or that methylation may only play a partial role in gene silencing in these cells (8).

Effect of pretreatment with 5-azacytidine on sensitivity to docetaxel in SKBR3 and Hs578t cells. (A) SKBR3 and (B) Hs578t treated with 1.25 μM 5-azacytidine (5-AzaC) for 72 h followed by 2.5 nM or 5 nM docetaxel treatment for 72 h. Apoptosis was assayed by annexin V/propidium iodide staining followed by flow cytometry.
Docetaxel is one of the commonly used chemotherapy drugs to treat breast cancer. The mechanism of action of docetaxel includes the binding and stabilizing of tubulin which results in cell cycle arrest hence inhibiting proliferation of cancer cells. In 1996, the Food and Drug Administration (FDA) approved its use to treat patients with locally advanced or metastatic breast cancer. In 2004, the FDA also approved docetaxel for adjuvant treatment in node-positive breast cancer in combination with doxorubicin and cyclophosphamide. However, around 50% of patients do not respond to docetaxel chemotherapy (14). Current studies are focused on identifying the molecular mechanisms responsible for docetaxel resistance in breast cancer (14-17). The down-regulation of pro-apoptotic genes by epigenetic modifications is one of the mechanisms of resistance of cancer cells to chemotherapeutic drugs. Recently, we showed that growth arrest and DNA damage inducible alpha (GADD45α), a gene involved in apoptosis, is deregulated by DNA methylation in prostate cancer and reactivation of gene expression by treatment with 5-azacytidine enhanced sensitivity of prostate cancer cells to docetaxel treatment (2). TMS1 is a pro-apoptotic gene that is down-regulated in breast cancer. The present results showed that pretreatment with 5-azacytidine up-regulated TMS1 expression and resulted in increased apoptosis after docetaxel treatment. This suggests that treatment with a demethylating agent such as 5-azacytidine prior to docetaxel may improve efficacy of treatment of breast cancer patients who are refractory to docetaxel treatment.
Acknowledgements
This work was supported by grants from the Women's Cancer Association of the University of Miami and Florida's Breast Cancer Coalition Research Foundation (R.S.).
- Received March 12, 2009.
- Revision received June 15, 2009.
- Accepted June 19, 2009.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- North American ATLL has a distinct mutational and transcriptional profile and responds to epigenetic therapies
- Phase I Study of CC-486 Alone and in Combination with Carboplatin or nab-Paclitaxel in Patients with Relapsed or Refractory Solid Tumors
- Role of DNA Methylation in Cabazitaxel Resistance in Prostate Cancer
- Targeting the epigenome and other new strategies in diffuse large B-cell lymphoma: beyond R-CHOP
- Mechanism-Based Epigenetic Chemosensitization Therapy of Diffuse Large B-Cell Lymphoma
- 5-Azacytidine Reverses Drug Resistance in Bladder Cancer Cells
- Methylation-mediated Silencing of TMS1 in Pancreatic Cancer and its Potential Contribution to Chemosensitivity