


Abstract
CVX-22 is a CovX-Body™, produced by covalently attaching a thrombospondin-1 (TSP-1) type 1 repeat peptide mimetic to a humanized IgG1 molecule. To dissect the antiangiogenic mechanism of CVX-22, the numbers and proliferative status of defined tumor endothelial cell (TEC) subsets from the B16 and C32 melanoma models were examined. CVX-22 treatment reduced the numbers of activated, vascular endothelial growth factor receptor 2 (VEGFR2)-positive TECs. Because the vast majority of mitotically active TECs reside in the VEGFR2 subset, a reduction in numbers of this compartment resulted in an 82% overall decrease in BrdU labeling of TEC. However, the rate of proliferation and VEGFR2 receptor density of this VEGFR2-positive subpopulation were unaffected. Instead, CVX-22 induced endothelial cell apoptosis both in vitro and in vivo, indicating that CVX-22 acts by selective deletion of activated, VEGFR2-positive TEC. The overrepresentation of activated cells in sites of tumor angiogenesis may confer a unique specificity of CVX-22 for tumor vasculature.
Thrombospondin-1 (TSP-1) is a multifunctional secreted glycoprotein that reduces angiogenesis and tumor growth in vivo, and induces endothelial cell apoptosis in vitro. TSP-1 has been identified as a tumor suppressor and its expression is lost in a wide variety of malignancies (1-5). Loss of TSP-1 expression is associated with increased tumor angiogenesis and microvessel density (1), and expression of TSP-1 correlates with improved prognosis in a variety of tumor types including colorectal, oral squamous cell, cervical, non small cell lung, pancreatic and melanoma (1-3, 5-8). Consistent with the strong correlation observed between TSP-1 loss and poor prognosis in human malignancies, transgenic models support a direct causal role for TSP-1 as a tumor suppressor, as increased tumor growth is observed in TSP-1 knockout mice (9). These data suggest that the broad prognostic advantage provided by TSP-1 is a result of antiangiogenic activity.
TSP-1 contains a number of functional motifs with distinct binding partners and functions, including three type 1 repeat domains. Antiangiogenic activity has been mapped to the thrombospondin type 1 repeat region, although other regions also play a role in TSP-1 antiangiogenic activity (10). The type 1 repeats of TSP-1 (TSR) have multiple known binding partners including CD36, TGF-Β, Β-1 integrin, heparan sulfate, and histidine-rich glycoprotein (11-15). Within the TSR domains, the sequence GVITRIR confers antiangiogenic activity, and TSR-derived peptides induce apoptosis and inhibit migration in a variety of normal primary endothelial cell populations including human umbilical artery endothelial cells (HUAECs), human umbilical vein endothelial cells (HUVECs), and human microvessel endothelial cells (HMVECs) (10, 16-18). Even small variations in the TSR sequence can alter the properties of the peptide. For example, a single amino acid change to a TSR peptide both increased inhibition of HMVEC tube formation, and decreased inhibition of HMVEC migration, suggesting multiple mechanisms of action, perhaps via multiple receptors or ligands (18).
The multiplicity of TSR ligands and receptors obscures the in vivo mechanisms of action and mechanism of specificity. Given this, it is difficult to predict the precise mechanism of action for TSR-derived peptides in vivo, yet tumor-specific antiangiogenic and antitumor effects are clear. TSR-derived peptides have shown anti-tumor activity in a wide variety of tumor models, including fibrosarcoma, A431 epidermoid carcinoma, ASPC1 pancreatic cancer, B16 murine melanoma, HT29 colon carcinoma, primary human bladder tumors, Lewis Lung carcinoma, PC-3 prostate adenocarcinoma, and others (16, 18-23). A decrease in microvessel density was described in many of these xenograft studies, although the cell type affected in vivo has not been defined (16, 21, 22, 24).
The in vitro induction of apoptosis of endothelial cells by TSR peptides might suggest a general induction of endothelial cell apoptosis in vivo as well, resulting in lower microvessel density. This general induction would imply that TSR peptides are vascular targeting agents as opposed to vascular normalizing or antiangiogenic agents. However, it is not clear which mechanism of action is employed by TSR peptides, nor if, or how, any specificity for tumor vessels would be provided. In the case of vascular endothelial growth factor (VEGF) inhibitors, the means of specificity for tumor vessels is clear, since tumor vessels overexpress VEGF receptor (25). However, the mechanism of action or specificity of TSR derivative drugs remains elusive, given the multiple possible effector molecules, and that a link between TSR activity and overexpression of a tumor ligand or receptor has not been described. To provide a drug with the antiangiogenic properties of TSP-1 and a more favorable pharmacokinetic profile, a TSR peptide mimetic was covalently linked to a humanized IgG1 antibody. The effects of the molecule, CVX-22, on tumor microvessel density and tumor volume, in view of its proposed antiangiogenic mechanism of action (26, 27), were investigated.
Materials and Methods
Drugs. CVX-22 was generated by the covalent attachment of the TSP-1 mimetic peptide CVX-1212 (Pro-Gly-Val-(D-alloIle)-Thr-Nva-Ile-Arg-Pro-NHEt) to the catalytic antibody CVX-02 as described elsewhwere (28). The purified peptide (3.0 molar equivalents) was combined with antibody CVX-02 (1.0 molar equivalent) in phosphate-buffered saline (PBS), pH 7.4 for 1 minute at room temperature to generate bivalent CovX-Body CVX-22. CVX-22 was dialyzed in a 10 MWCO slidealyzer (Pierce, Rockford, Ill., USA) into PBS overnight with 2x buffer changes in order to remove unconjugated peptide. The reaction was monitored by 316 nm spectral absorbance normalized to protein concentration as measured by 280 nM absorbance (A280). Conjugation was verified by UV spectrometry and size exclusion chromatography-high pressure liquid chromatography (SEC-HPLC). One mg/ml aliquots of CVX-22 in PBS were stored at -20°C until use. PBS was used as a control agent. CVX1411, a control non-targeting CovX-Body, is composed of the catalytic antibody CVX-02 and a chemical linker as for CVX-22, but with a covalently attached valine instead of the TSP-1 peptide.
Pharmacokinetics. The pharmacokinetics of CVX-22, and the free TSP-1 peptidomimetic CVX-1212 were determined in male Swiss Webster mice (Charles River Laboratories, Wilmington, MA, USA). Mice were dosed with compounds at 10 mg/kg intravenously (i.v.), and blood samples from three mice per timepoint were taken 0.08, 0.25, 0.5, 1, 2, 4, 6, and 24 hours, and serum samples prepared by centrifugation. CVX-22 concentrations were determined using a rabbit polyclonal antibody generated against the TSP-1 peptidomimetic portion of CVX-22 in a capture ELISA. In brief, plates were coated with anti-peptidomimetic antibody, and serum samples added. The Fc portion of CVX-22 was then detected by addition of HRP-conjugated donkey anti-human IgG (Bethyl Laboratories, Montgomery, TX, USA), and TMB developing solution (Kirkegaard and Perry Laboratories, Gaithersburg, MD, USA). CVX-1212 concentrations were determined by LC/MS. Data were analyzed using WinNonlin (version 5.0.1, Pharsight Corp., Mountain View, CA, USA).
In vivo tumor models. Adult female nu/nu mice (Charles River Laboratories strain NU-Foxn1nu, 6 weeks, 20-25 g) and adult female C57BL/6 mice (Charles River Laboratories, 6 weeks, 20-25g) were housed 10 per cage in a temperature- and light-controlled vivarium (light/dark 12 h: 12 h) with standard laboratory chow and water available ad libitum. Mice were allowed to rest undisturbed for at least 48 hours after arrival before experimentation was initiated. All animal experiments were conducted after protocol approval by the CovX Institutional Animal Care and Use Committee.
B16 murine melanoma was chosen for its historical response to TSP-1-based drugs, while C32 was chosen as a human melanoma counterpart. B16-F10 murine melanoma (American Type Culture Collection, Manassas, VA USA; CRL-6375) and C32 human melanoma (American Type Culture Collection; CRL-1585) cell lines were cultured in RPMI-1640 medium with 10% fetal bovine serum (FBS). Sub-confluent tumor cells (70-80%) were harvested after brief treatment with 0.25 % trypsin (Cellgro, Mediatech Inc, VA) and resuspended in Hank's balanced salt solution (HBSS) for inoculation. Mice were injected with 3×106 C32 cells in a total 0.1 ml HBSS + 0.1ml Matrigel (BD Biosciences, San Jose, CA USA; cat #35423) and C57BL/6 mice were injected with 0.5×106 B16 F10 cells in a total of 0.2 ml of HBSS. Tumor cell suspensions were injected subcutaneously (s.c.) into the right flank of each mouse.
Treatments were initiated the day after tumor inoculation. CVX-22 was injected twice weekly (10 mg/kg i.v.). All treatments were administered in a volume of 0.2 ml/mouse, and conducted in conscious animals using brief manual restraint. Treatment was continued for two weeks for the B16 study, and tumors harvested while relatively small to facilitate FACS analysis. Treatment was continued for four weeks for the C32 study. A second identical B16 study was continued for 3 weeks, at which time tumors were harvested for immunohistochemistry. Tumor measurements were made with manual calipers twice weekly and tumor volumes were calculated using the formula 0.526 × length × width2. Osmotic pumps with a flow rate of 8 μl/hour (Alzet, Cupertino, CA USA; #2001D) containing 3 mg/ml BrdU (BrdU Flow Kit; BD Pharmingen, San Diego, CA USA; Cat #559619) were implanted subcutaneously 48 hours after a final dose of drug. Animals were sacrificed 24 h after pump implant. Tumors were excised at the end of the study, weighed, and processed for immunohistochemistry or FACS analysis.
Flow cytometric analysis of tumors. Tumors were minced in 10 ml disaggregation mixture (HBSS, 1% bovine serum albumin (Sigma Aldrich, St. Louis, MO, USA; Cat #A7030), collagenase type 2 (Invitrogen, Carlsbad, CA, USA; Cat #17101-015) and collagenase type 4 (Invitrogen; Cat # 17104-019), final concentration 250 units/ml) for FACS analysis. Disaggregated tumors were incubated rotating at 37°C for 1.5-2 h, and digestion stopped by adding 5 ml cold FBS (Invitrogen). Tumors were disaggregated by Medimachine (BD Biosciences; Cat #340587), and filtered through a 50 μM screen. Cells were collected by centrifugation at 1500 RPM at 4°C for 5 minutes, resuspended in 5 ml red blood cell lysis buffer (Sigma Aldrich; Cat # R7757), incubated 5 minutes on ice, washed twice in PBS + 20% FBS, counted and resuspended at 1×106 cells per 100 μl in cold FACS resuspension buffer (PBS + 20% FBS, 5% BSA, 10 μg/ml mIgG (Pierce; Cat #31204). One hundred μl of cells were used per staining reaction.
Cells from each tumor were stained with antibodies including FITC-conjugated anti-mouse CD31 (BD Pharmingen; Cat #553372), PE-conjugated anti-mouse VEGFR2 (BD Pharmingen, Cat #555308), AlexaFluor-647-conjugated anti-BrdU-(Invitrogen; Cat #A21305) and 7AAD (BD Pharmingen; Cat #559925) in a four-color stain. In addition, cells were stained with PE-conjugated anti-mouse CD31 (BD Pharmingen; Cat # 553373) and FITC-conjugated anti-mouse CD117 (BD Pharmingen; Cat # 553354) for enumeration of endothelial progenitor cells; or FITC-conjugated anti-human CD44 or anti-mouse CD44 (BD Pharmingen; Cat #553133) for enumeration of C32 or B16 melanoma cells, respectively. Isotype controls and single-color stains were included as controls for flow cytometry settings and analysis. Ten μl of each antibody were added to 100 μl of cells, and tubes were incubated on ice for 45 minutes. Cells were then washed in 3 ml PBS + 20% FCS. For BrdU staining, cells were stained with the BrdU flow kit (BrdU Flow Kit, BD Pharmingen; Cat #559619) per manufacturer's instructions. Data was acquired on a BD LSRII, with analysis via FloJo (v6.4.7, FloJo, LLC, Ashland, OR, USA). Data was acquired for 100,000 cells from each sample, and all samples were autocompensated via FloJo.
Microvessel density and BrdU labeling. Snap-frozen tumor sections were oriented and embedded in optimum cutting temperature compound (OCT, Tissue-Tek; Proscitech, Kirwan, AUS) blocks. Three 5 μm step sections, cut one section every 150 μm, were obtained from each block using a cryostat. The slides were immediately fixed in cold acetone for 2 minutes and air dried, then processed for double immmunofluorescence staining of BrdU and CD31. Slides were first treated with 2 N HCl for 30 minutes at room temperature to denature DNA. Slides were then washed twice with PBS and treated with Autofluorescence Eliminator Reagent (Chemicon; Cat# 2160) for 2 minutes. Sections were incubated overnight at 4°C with primary antibodies against BrdU (BD Pharmingen; FITC conjugated mouse Anti-BrdU, cat # 347583, diluted 1:10) and mouse CD31 (BD Pharmingen; Rat anti-mouse CD31, Cat #550274, diluted 1:50). Slides were then treated with AlexFluor 555 conjugated goat anti-rat IgG (Invitrogen; Cat # S32355, diluted 1:500) for detection of CD31, incubated at room temperature for 1 hour. Sections were rinsed with PBS between each step. After staining, slides were mounted with Antifade and DAPI (Invitrogen; cat# 24636). CD31-and BrdU-positive areas in each tumor were imaged using a Qimaging Micropublisher 5.0 RTV camera coupled with a Nikon Eclipse 80i fluorescence microscope equipped with Tritc/rhodamine, FITC and DAPI filter cubes respectively at each x20 microscopic field. Five evenly spaced images were taken along a cross section of each tumor, starting and ending at a point on the viable rim in which CD31 staining was greatest. The collected images were then processed and analyzed with ImagePro 5.1 software (Media Cybernetics; Bethesda, MD, USA). CD31 and/or BrdU-positive pixels/per x20 microscopic field were calculated by dividing the total CD31-positive area of the three sections of each tumor by 15 (five fields from each of three sections from each tumor).

Pharmacokinetics of CVX-22 and CVX-1212 in the mouse. Mice were dosed with CVX-22 and CVX-1212 at 10 mg/kg i.v. Values are expressed as averages from three mice±the standard error (SEM).
Quantification of apoptosis in tumors. Double immmunofluorescence staining of cleaved caspase-3 and CD31 was carried out on five frozen sections (5 μm thickness) of each B16 tumor. Briefly, slides were stained with a cocktail of primary antibodies against cleaved caspase-3 (Cell Signaling Technology, Danvers, MA, USA; Cat #9664 Rabbit mAb, diluted 1:100) and mouse CD31 overnight at 4°C, after treatment with Autofluorescence Eliminator Reagent (Chemicon; Cat# 2160) for 2 minutes, streptavidin-biotin block (Vector Lab. Burlingame, CA, USA; cat #SP-2002) for 30 minutes and nonspecific staining block with 2.5% bovine serum albumin (BSA) for 15 minutes. The second antibody for cleaved caspase-3 was biotinylated goat anti-rabbit IgG (Vector Laboratory; cat#PK-6101, diluted 1:200) which was incubated at room temperature for 1 hour. The slides were then treated with a cocktail of AlexFluor 555 conjugated Strepavidin (Invitrogen; cat #S32355, diluted 1:400) for detection of cleaved caspase-3, and FITC-labeled polyclonal anti-rat IgG for detection of CD31, both of which were incubated at room temperature for 1 h. Sections were rinsed with PBS between each step. After staining, the slides were mounted with Antifade with DAPI. Three fluorescence images of cleaved caspase-3, CD31 and nuclei from each section were collected using a Qimaging Micropublisher 5.0 RTV camera coupled with a Nikon Eclipse 80i fluorescence microscope equipped with Tritc/rhodamine, FITC and DAPI filter cubes, respectively at each x20 microscopic field. The collected images were then processed, merged and analyzed using ImagePro 5.1 software. Quantification of apoptosis was calculated as cleaved caspase-3 immunofluorescence pixels per x20 field.
In vitro apoptosis. HUVEC (BD Biosciences) were seeded at a density of 10,000 cells per well in 8-well chamber slides (BD Falcon, BD Biosciences). After 24 hours, PBS, CVX-22, and CVX1411 were added at concentrations of 0.5 uM, 5 uM and 10 uM. Cells were cultured for an additional 24 hours. Cleaved caspase-3 was detected with anti-cleaved caspase-3 (Santa Cruz Biotechnology, Santa Cruz CA, USA, Cat #22-171 at a dilution of 1:100), and Alexa Fluor 555-conjugated goat anti-rabbit IgG (Invitrogen) diluted 1:400. Cleaved caspase-3 stained slides were mounted with DAPI containing anti-fade mounting medium. Three photos were captured per well in areas of maximal staining for cleaved caspase-3 with a Qimaging Micropublisher 5.0 RTV camera coupled with a Nikon Eclipse 80i fluorescence microscope. The collected images were then processed, merged and analyzed using ImagePro 5.1 software. Quantification of apoptosis was calculated as total cleaved caspase-3-positive objects per x40 microscopic field divided by the number of DAPI-positive nuclei.
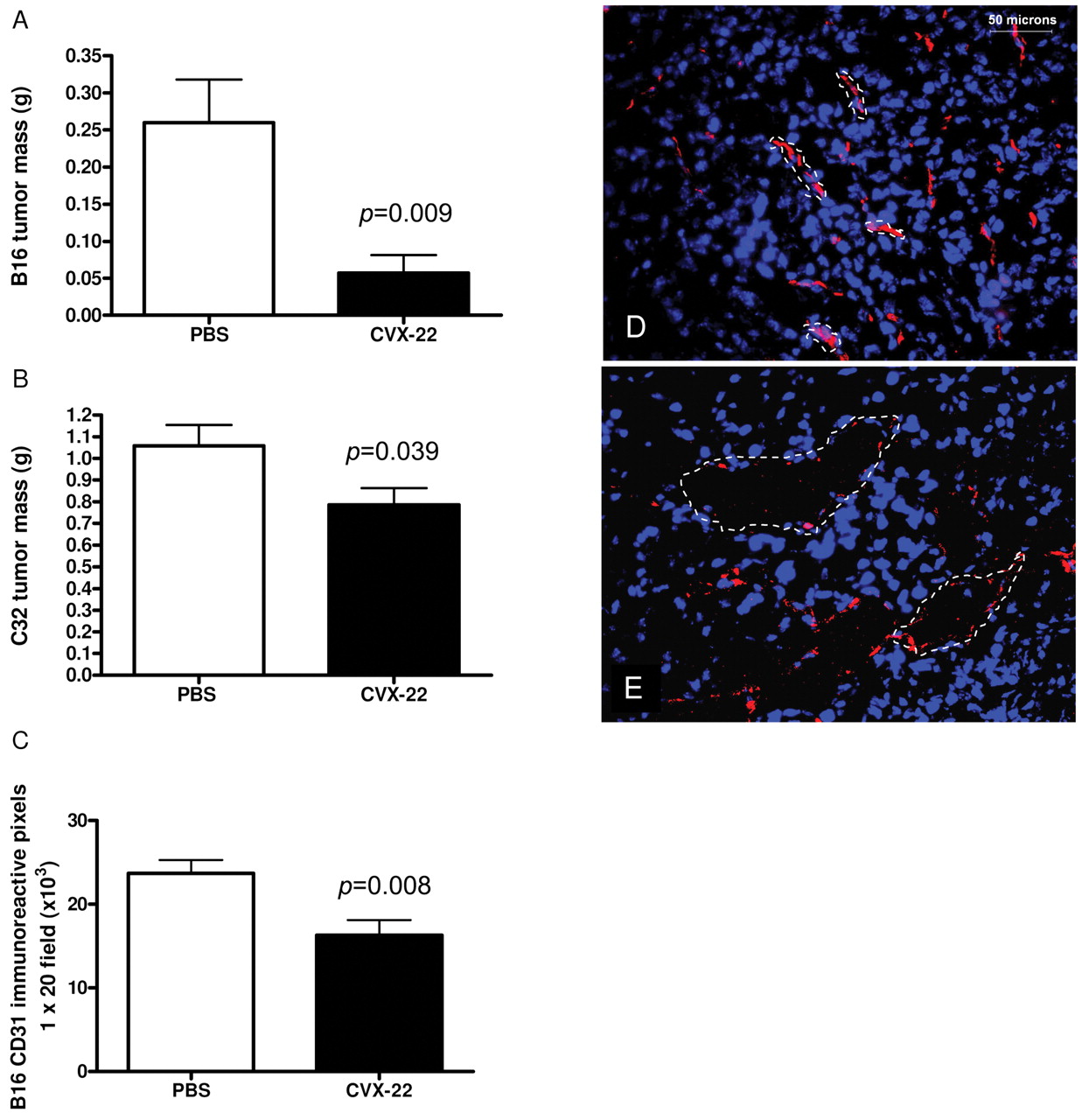
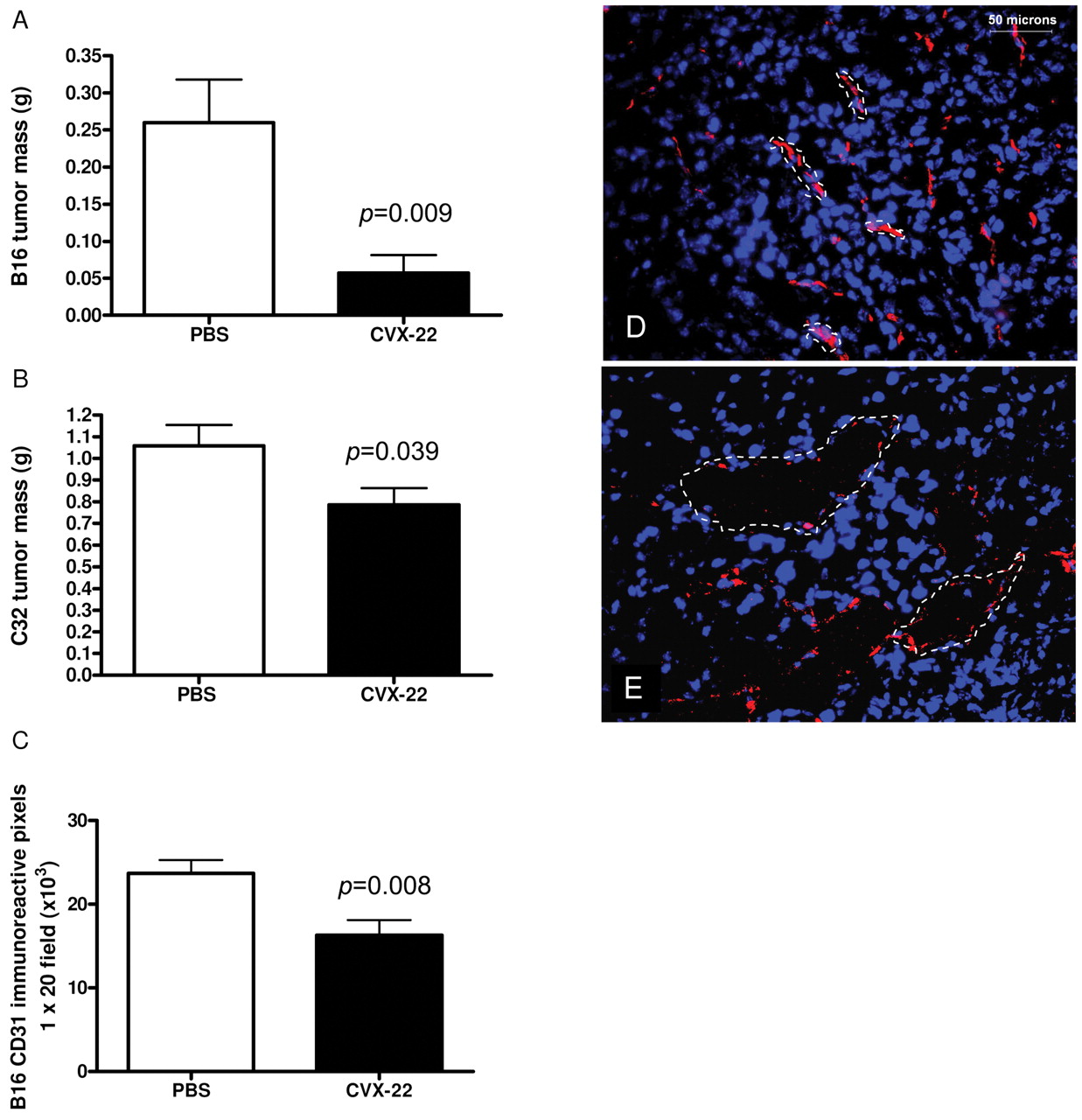
CVX-22 reduced tumor growth in B16 murine melanoma (A) and in C32 human xenograft melanoma (B) models. Data are depicted as means±SEM; n=10 animals/group. B16 tumors were allowed to grow for 2 weeks and C32 tumors for 4 weeks. At this time, the studies were terminated and tumors excised and weighed. Microvessel density was reduced in B16 tumors (C). Morphology of tumor vessels (dotted lines) was altered by CVX-22, showing enlarged lumens lined by disconnected endothelial cells (CD31, red). Nuclei in blue (DAPI). PBS-treated tumors (D), CVX-22-treated tumors (E).
Data analysis. All data were assessed in GraphPad Prism (version 4.00 for Windows; GraphPad Software, San Diego California USA) for statistical significance by two-tailed t-test or ANOVA with Dunnett's post-hoc analysis, and depicted as means±SEM.

A significant decrease in BrdU labeling in CD31+ tumor endothelial cells treated with CVX-22, as measured by immunohistochemistry (A). A decrease in BrdU labeling was observed in B16 tumor cells treated with CVX-22 (B). Data are depicted as means±SEM; n=10 animals/group.
Results
CVX-22 pharmacokinetics in mouse. The half-life of the peptidomimetic CVX-1212 was estimated to be approximately 0.3 hours using a one-compartment model, and peptide levels were below detection limits 4 hours after i.v. dosing (Figure 1). In contrast, CVX-22 had an estimated alpha phase half-life of 0.2 hours, a beta phase half-life of ~3.4 hours and an area under the curve (AUC) of 44.2 μg h/ml, far in excess of what has been reported for other TSP-1 peptidomimetics in vivo (18). These data demonstrate the profound pharmacokinetic benefit of covalently fusing the TSP-1 peptidomimetic to the CVX-02 IgG1.
CVX-22 reduced tumor mass and microvessel density (MVD). CVX-22 at 10 mg/kg twice weekly inhibited tumor growth in the B16 syngeneic and C32 human xenograft models of melanoma (Figure 2). B16 tumors were allowed to grow for 2 weeks and C32 tumors for 4 weeks. At this time, the studies were terminated and tumors excised and weighed. The average B16 tumor mass was reduced by 78% by CVX-22 treatment (Figure 2A). To confirm that the effects were not specific to the syngeneic B16 model, the C32 xenograft model was also examined. The average C32 tumor mass was reduced 26% by CVX-22 (Figure 2B). Flow cytometry consumes the entirety of the tumors employed and a second study was run for immunohistochemistry of B16 but not C32 tumors. Consistent with its proposed role as an antiangiogenic drug, CVX-22 reduced MVD by 32% in B16 tumors (Figure 2C). The morphology of microvessels in B16 tumors treated with CVX-22 was also dramatically altered, showing enlarged and thin-walled lumens, lined by disconnected endothelial cells (Figure 2D, E).
CVX-22 reduced the proliferation index of tumor endothelial cells (TEC) and tumor cells. The overall proliferative index of B16 TECs was significantly reduced by treatment with CVX-22, reflected by an 83% decrease in BrdU labeling in CD31-positive cells as measured by immunohistochemistry (Figure 3A). BrdU labeling of B16 tumor cells was reduced by 68% by CVX-22 treatment (Figure 3B), although the presence of a low-labeling outlier in the PBS-treated animals obviated the statistical significance of this effect.
CVX-22 reduced the number of VEGFR2-positive TECs. The observed reduction in the number of TECs that proliferated in a 24-hour period could have resulted from a diminution in the global rate of proliferation, or could reflect a numerical reduction in the pool of cells that actively undergo proliferation. To distinguish between these two possibilities, TECs and subsets there of were enumerated through flow cytometry. In a representative PBS-treated tumor, ~64% of events were selected to maximize the number of TECs counted by excluding low forward scatter/side scatter (FSC/SSC) debris and high FCS/SSC aggregates (data not shown), and designated R1. One hundred thousand R1 events were counted for each sample. TEC were then selected by serial gating on the CD31-FITC-positive cells (R2), which comprised ~17% of R1 cells. TECs that were positive for VEGFR2 were identified by serial gating of VEGFR2-PE stain (R3), which comprised ~15% of R2 cells. Proliferation during the 24 hours prior to tumor harvest, during which time a BrdU osmotic pump was present, was assessed by anti-BrdU-Alexa-Fluor647 stain.
Treatment with CVX-22 reduced the proportion of TECs that were VEGFR2-positive from both B16 and C32 tumors (Figure 4). In PBS-treated B16 tumors, 14.5% of TEC were positive for VEGFR2 by FACS, while treatment with CVX-22 reduced the percentage of VEGFR2-positive TEC to 7.5%, a significant 48% reduction (Figure 4A). In PBS-treated C32 tumors, the proportion of cells that were positive for VEGFR2 was 54% in this highly vascular tumor. CVX-22 treatment lowered the C32 VEGFR2-positive population to 42% of the TEC pool, a significant 21% reduction (Figure 4B). To control for artifacts of TEC preparation from tumors, numbers of VEGFR2-negative TECs were quantified as a proportion of the total cell preparation. The percentage of VEGFR2-negative cells in the cell preparations was unchanged in all treatment groups in both B16 and C32 tumors (Figure 4 C, D). This validates the reduction of VEGFR2-expressing TECs as an effect specific to that subpopulation of cells.

CVX-22 treatment reduced numbers of VEGFR2+ TEC in B16 tumors (A), and in C32 tumors (B) as measured by flow cytometry. Data are depicted as means±SEM; n=10 animals/group.
The VEGFR2-positive subset of TEC is largely responsible for neoangiogenesis, and VEGFR2 is a well-validated marker of activated endothelial cells (29). Fifteen percent of all TECs in PBS-treated tumors were VEGFR2+ (Figure 4A, B). In PBS-treated tumors, 23% of this VEGFR2+ population labeled with BrdU in 24 hours, reflecting robust growth of these cells (Figure 5A). This VEGFR2/BrdU double-positive population thus comprises 3.5% of all CD31+ TEC (15% × 23% =3.5%). In contrast, only 5% of the entire TEC population was labeled with BrdU (Figure 5A). This indicates that 70% of all TECs in mitosis are VEGFR2-positive (3.5% out of 5%), despite the fact that VEGFR2+ cells comprised only 15% of TEC. Thus, a reduction in numbers of VEGFR2+ TECs resulted in a reduced number of BrdU+ TECs, as observed. This is also apparent in Figure 3, where a reduction in BrdU labeling was measured by IHC. Because VEGFR2 expression is also a marker of endothelial progenitor cells (EPC), it was possible that a reduction in this population might represent EPC depletion. The CD31/CD117 double-positive population is inclusive of EPC (30), and was quantified in C32 tumors. Numbers of these cells were similar in all treatment groups, with 19% ± 0.7 (mean±SEM) of all CD31-positive cells also expressing CD117, suggesting that EPC numbers were unaffected by CVX-22 (data not shown).
Induction of endothelial cell apoptosis by CVX-22. A reduction in the VEGFR2-positive population of cells might result from one of three possible mechanisms. First, the expression levels of VEGFR2 might be reduced. Second, the proliferation rate of the VEGFR2-positive cells might be reduced by CVX-22, resulting in a net decrease in the numbers of progeny cells. Third, the VEGFR2-positive cells might be removed by a process of attrition such as apoptosis. To distinguish between these possibilities, VEGFR2 expression levels and level of BrdU labeling of this population was examined in B16 tumors. A marginal reduction in VEGFR2 expression levels was observed here, suggesting a slight reduction in receptor density (Figure 5B). It is possible that this effect made a minor contribution to the reduction in VEGFR2-positive TEC observed. However, other studies showed nonsignificant increases of similar magnitude, or no change (data not shown). Next, BrdU labeling was examined in VEGFR2-positive cells and showed no significant change between PBS- and CVX-22-treated groups (Figure 5C). These data indicate that the rate of mitosis of the VEGFR2-positive cell subset was unchanged by treatment with CVX-22. Taken together, the data indicate that the most likely mechanism for reduction of the VEGFR2-positive population by CVX-22 is accelerated cell death. To examine this, cleaved caspase-3 levels were examined by immunohistochemistry of B16 tumors (Figure 6A, B). An increase in apoptotic TECs was readily observed in tumors treated with CVX-22 in comparison to PBS-treated tumors. However, the difference between treatment groups was not statistically significant due to the presence of outliers (data not shown).
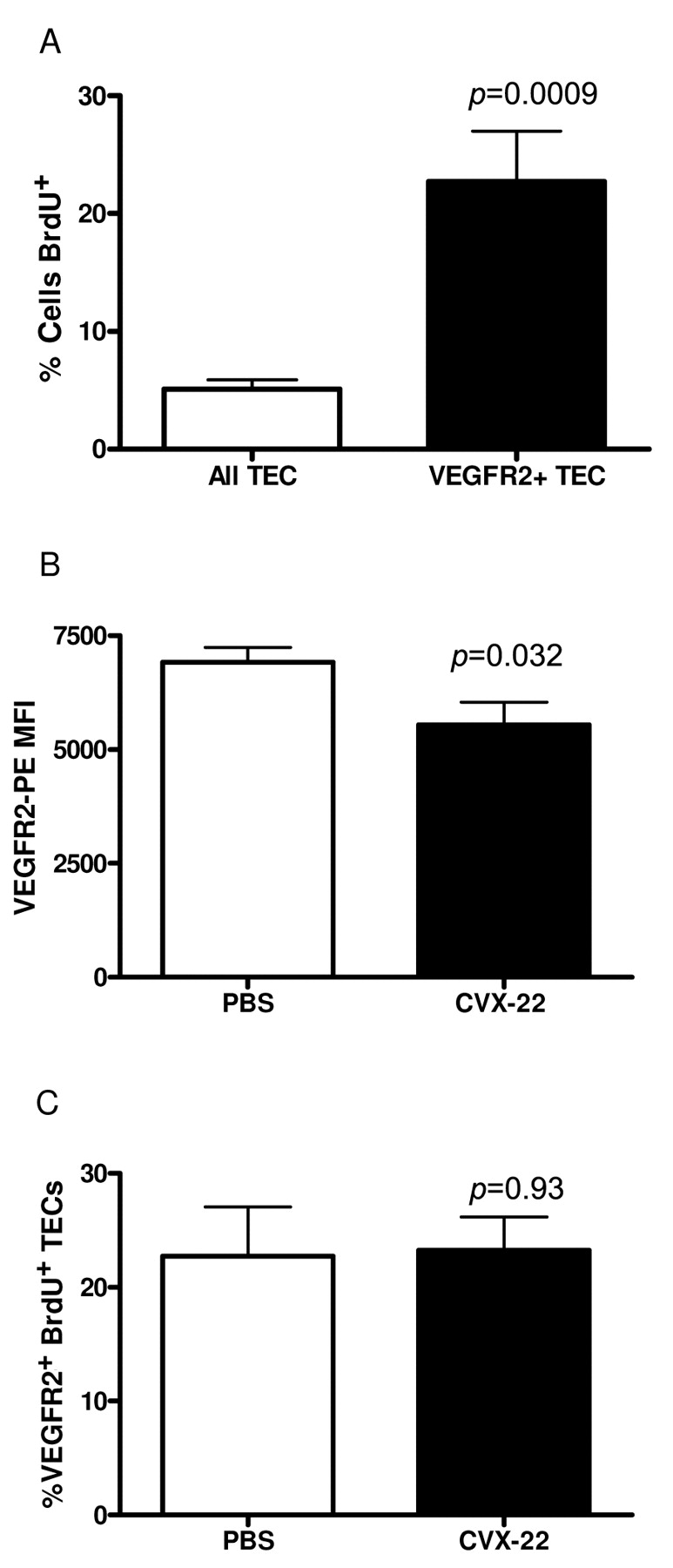
The proliferating pool of TECs resides in the VEGFR2+ subset in B16 tumors. Of all TEC, 5% were labeled with BrdU in vivo vs. 23% labeling for VEGFR2+ TECs as measured by FACS (A). CVX-22 reduced TEC VEGFR2 expression levels as measured by mean fluorescence intensity (MFI) of stain by FACS (B). CVX-22 treatment did not decrease BrdU labeling of VEGFR2+ TECs (C). Data are depicted as means±SEM; n=10 animals/group.
To confirm a direct apoptotic effect on endothelial cells, cultured HUVECs were treated with CVX-22, and apoptosis measured by levels of cleaved caspase-3, and proliferation by BrdU labeling (Figure 6C). Consistent with what was observed in vivo, endothelial cell apoptosis was induced by CVX-22, while proliferation rates remained unchanged (data not shown). Cells treated with 10 μM negative control CVX-1411 showed levels of apoptosis not different from those for PBS-treated cells at both 24 and 48 hours, while cells treated with 10 μM CVX-22 showed a dramatic induction of apoptosis, with 38% and 61% of all HUVECs staining for caspase-3 at 24 and 48 hours of treatment, respectively. Cells treated with 0.5 μM and 5 μM CVX-22 showed a 12% and 30% increase in apoptosis, respectively.
Discussion
As an antibody covalently linked to a TSP-1 peptidomimetic, CVX-22 represents a new class of antiangiogenic drugs. In this manuscript, we describe the population of cells targeted by CVX-22, resulting in an antiangiogenic effect in vivo. A thorough understanding of the cellular action of novel antiangiogenics is crucial to forming rational therapeutic strategies and increasing the probability of benefit to patients while minimizing risk. In the case of TSR-derived drugs, the agents may operate through multiple possible receptors in vivo, and may have both agonist and antagonist activities, obscuring the most clinically relevant actions. In particular, the mechanism by which these agents might have specificity for tumor vasculature remains unexplained, as tumor-specific overexpression of a ligand or receptor related to TSR activity has not been described, and the cell population affected in vivo has not been previously identified.

Induction of apoptosis in tumor and cultured endothelial cells by CVX-22. Representative cleaved caspase-3 immunoreactivity in TECs from B16 tumors treated with PBS (panel A), or CVX-22 (panel B). Green: CD31; red: cleaved caspase-3; blue: DAPI; orange: overlay of CD31 and cleaved caspase-3 (arrows). Increased cleaved caspase-3 staining in HUVEC treated for 24 hours with CVX-22 (C). Cells were treated with PBS, CVX-22 or CVX-1411, a nontargeting CovX-Body™. *p<0.05, **p<0.01.
In this report, we described a novel mechanism of specificity for tumor vasculature for CVX-22, a TSR peptide mimetic-antibody fusion with favorable pharmacokinetic properties in comparison with our TSP-1 peptidomimetic, and with previously examined TSP-1-derived peptides (18). By employing standard in vivo tumor models in combination with ex vivo analysis of TEC subsets, we demonstrated that CVX-22 exerted an antiangiogenic effect by eradicating activated TECs. CVX-22 reduced tumor growth in two models of melanoma, and reduced microvessel density, consistent with the proposed antivascular role of CVX-22. We observed that BrdU labeling of TECs was dramatically reduced by treatment with CVX-22. However, rates of cell division for individual TEC subpopulations were unchanged. Instead, the most active cells were eradicated, resulting in a net loss of labeled TECs. As has been abundantly described elsewhere, VEGFR2 is an excellent marker for activated TECs, and we observed that this specific subpopulation was reduced by CVX-22 treatment. This degree of specificity may contribute to the favorable toxicity profile of TSP-1 peptides in human trials (31-34).
First, it was confirmed that VEGFR2 served as a surrogate marker for active TECs, as the majority of the mitotic activity of TECs was contained within this subset. The mitotically active VEGFR2-positive subset of TEC was selectively reduced by CVX-22 treatment. Furthermore, reduction in numbers of VEGFR2-positive cells was the result of apoptosis as opposed to induction of quiescence or receptor down-regulation. Although the molecular mechanism underlying the induction of apoptosis by TSP-1 mimetic peptides has been obscured by the complexity of TSP-1 biology, we demonstrate here that the effect has specificity for the most activated TEC subset.
In this study, VEGFR2 served as a surrogate marker for activated TEC, rather than as a target for our drug. Because VEGFR2 induces proliferation and is coexpressed with other markers of activated TEC including ANGPT2 and VEGFR1 (FLT1), it serves as a useful marker for proliferating TECs (29). In contrast to VEGFR2-positive TECs, little or no cell division is observed in normal endothelial cells in the absence of wound healing (35). Because endothelial cell division is required for angiogenesis, targeting of active TECs by cytotoxic chemotherapy has an antiangiogenic effect (36). Further, it has been clearly demonstrated with many agents that inhibition of activated TECs directly by VEGF or VEGFR2 inhibitors blocks angiogenesis and decreases tumor growth. Our data suggest that CVX22 also targets this population, providing specificity for tumor vasculature.
Peptides and monoclonal antibodies have emerged as important therapeutics, but each still have many limitations. Peptides are highly potent, but frequently are poor therapeutics due to rapid degradation in the body and required frequency of administration. Traditional monoclonal antibodies have more favorable pharmacokinetics, but are often constrained by a challenging and lengthy development process. CVX-22 is based on a novel approach that combines the strengths of both peptides and antibodies into a new molecule. The net result is a peptide-containing drug that produced tumor responses in animals with doses over 150-fold lower on a molar basis than previously reported for free peptide TSP-1-based drugs (18).
Many new antiangiogenic drugs are entering the clinic, while our knowledge of the mechanism of action of these drugs is only just emerging. However, knowledge of the mechanism of action is a prerequisite to formulation of rational combination strategies, especially as it is unlikely that any antiangiogenic agent will optimally control tumor growth in a clinical setting as a single agent. Our demonstration of the selective eradication of active TECs by CVX-22 may likewise allow more strategic clinical approaches in the future. For example, combinations with drugs targeting non-overlapping cell subsets such as vessel pericytes may provide additive benefit.
Acknowledgements
We would like to thank Rodney Lappe, Curt Bradshaw and Gary Woodnutt for leadership and guidance during this study. We would also like to thank the chemists of CovX including Venkata Doppalapudi, David Tumelty, John Rizzo, and Jing-Yu Lai for production of the drug components described in this study.
- Received June 26, 2008.
- Revision received January 14, 2009.
- Accepted February 18, 2009.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}